
Enzyme-free selective determination of H2O2 and glucose using functionalized CuNP-modified graphite electrode in room temperature ionic liquid medium†
Abstract
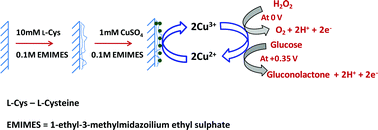
A novel enzyme-free modified electrode constructed by electropolymerization of L-cysteine on a graphite electrode, followed by electrodeposition of copper nanoparticles using ionic liquid as a green electrolyte. The modified electrode exhibits an excellent electrocatalytic activity towards the oxidation of hydrogen peroxide (H2O2) and glucose at a reduced overpotential of 0 V and +0.35 V, respectively. The immobilization of copper nanoparticle encapsulated by polycysteine (PC) formed on the electrode was confirmed by Fourier transform infrared (FT-IR) spectroscopy, Raman spectra, X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM) and atomic force microscopy (AFM) studies. The determination of H2O2 and glucose with the modified electrode shows the advantages of ease of preparation, high sensitivity, good selectivity and stability. The practical application of the modified electrode for selective detection of H2O2 and glucose has been evaluated by analyzing the real samples of stain remover solutions and human urine samples to determine H2O2 and glucose, respectively.
 Please wait while we load your content...
Please wait while we load your content...

