
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new class of pyrenyl solid-state emitters: 1-pyrenyl ynones. Synthesis via the Friedel–Crafts route, molecular and electronic structure and photophysical properties†
Rafał Flamholca,
Damian Plażuka,
Janusz Zakrzewski*a,
Rémi Métivier*b,
Keitaro Nakatanib,
Anna Makalc and
Krzysztof Woźniakc
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland. E-mail: janzak@uni.lodz.pl
bPPSM, ENS Cachan, CNRS, UniverSud, 61 av President Wilson, 94230 Cachan, France. E-mail: metivier@ppsm.ens-cachan.fr
cDepartment of Chemistry, Warsaw University, Pasteura 1, 02-093 Warszawa, Poland
First published on 15th July 2014
Abstract
Friedel–Crafts acylation of pyrene with alkynoic acids in the presence of trifluoroacetic anhydride and triflic acid constitutes a direct and efficient route to 1-pyrenyl ynones. These compounds in chloroform solution emit fluorescence at longer wavelengths, with higher quantum yields and longer lifetimes than a typical saturated acylpyrene derivative, 1-acetylpyrene. Moreover, in contrast to 1-acetylpyrene, they are moderate solid-state emitters. Comparative DFT studies revealed strong stabilization of the LUMOs of 1-pyrenyl ynones in comparison to the LUMO of 1-acetylpyrene. The single-crystal X-ray structure of 1-(pyren-1-yl)but-2-yn-1-one showed π-interactions of pyrenyl moieties in the crystal lattice. Investigations of the solid-state fluorescence of this compound revealed emission from long-lived excited states, including excimer species.
Introduction
Pyrene and its derivatives have attracted considerable interest as materials for organic electronics,1 fluorescent monomers,2,3 molecular probes,4–7 and sensors.8,9 Considerable effort has been focused on the development of synthetic routes to pyrenes bearing different substituents1,10–12 and on studies of their fluorescence properties.13,14 Special attention has been paid to 1-pyrenyl carbonyl compounds such as aldehyde, ketones, acids, esters, amides, etc., exhibiting strongly environment-sensitive fluorescence and used as molecular probes.15–21 Recent works by the Konishi group have provided a basis for understanding the photophysical properties of this class of fluorophores.15–17,21 As far as pyrenyl ketones are concerned, research reports have mainly concentrated on pyrenyl alkyl ketones. This class of perylenyl fluorophores shows short fluorescence lifetimes (10−9–10−8 s, compared with >10−7 s for pyrene) and low quantum yields due to efficient intersystem crossing.14,15 Only very recently have two reports appeared disclosing the synthesis and photophysical properties of the simplest pyrenyl alkynyl ketone, 1-propynoilpyrene.22,23 This ketone offers a unique opportunity for attachment of a pyrene carbonyl tag to biomolecules or nanomaterials via azide-alkyne click chemistry.In a continuation of our research programme, which has focused on the use of functionalised carboxylic acids as acylating agents in Friedel–Crafts reaction, we recently elaborated an efficient method of synthesis of ferrocenyl ynones via a direct reaction of ferrocene with alkynoic acids in the presence of trifluoroacetic anhydride (TFAA) and trifluoromethanesulfonic acid (TfOH).24 Herein we report that this approach may be used for simple and efficient synthesis of 1-pyrenyl ynones. We have also studied the fluorescence properties of these compounds which were compared to those of the simplest pyrenyl alkyl ketone, 1-acetylpyrene. Unexpectedly, we found that, in contrast to the latter compound, the synthesized 1-pyrenyl ynones display fluorescence not only in solution but also in the solid state. An X-ray diffraction study (including topological analysis of experimental charge density) performed for 1-(pyren-1-yl)but-2-yn-1-one revealed face-to-face π-stacking of the pyrene moieties in the crystal, thus suggesting that emission may originate from solid-state excimers. Finally, we carried out comparative DFT calculations on this compound and 1-acetylpyrene.
Results and discussion
Synthesis of pyrenyl ynones 1a–d
Ynones or α,β-acetylenic ketones are versatile building blocks in organic synthesis, especially in the synthesis of heterocyclic compounds.25 They can be synthesized via various routes starting from terminal alkynes.26–29 Readily available 2-alkynoic acids30 are another potential starting material in ynone synthesis via Friedel–Crafts acylation of arenes. We reported the first example of such reaction, in which ferrocene was used as a reactive π-rich arene.24 Since pyrene also exhibits high reactivity towards electrophiles, it seemed interesting to evaluate whether this approach can be used for a direct introduction of the alkynoyl group into this polycyclic arene.The Friedel–Crafts reaction of pyrene with 2-alkynoic acids (Scheme 1) was carried out under conditions described earlier for ferrocene.24 The isolated yields of compounds 1a–c were in the range of 69–74%. Similarly, as in the case of ferrocene, the reaction of pyrene with propiolic acid (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H) led to an intractable reaction mixture. However, compound 1d was prepared in an almost quantitative yield (99%) by fluoride-promoted desilylation of 1c.
H) led to an intractable reaction mixture. However, compound 1d was prepared in an almost quantitative yield (99%) by fluoride-promoted desilylation of 1c.
 | ||
| Scheme 1 Synthesis of 1-pyrenyl ynones 1a–d. | ||
Structures of synthesized ynones 1a–d were confirmed by spectroscopic and elemental analysis data (see ESI†). The simplest compound of this series, 1d, was already reported in the literature. It was synthesised 40–44% overall in a reaction of pyrene-1-carboxaldehyde with ethynyl magnesium bromide22 (or TMS-ethynyl magnesium bromide followed by desilylation23), and subsequent oxidation of the alcohol formed with Jones reagent. Compared to those methods our synthesis was simpler (using pyrene as the starting material) and more efficient (71% overall yield).
Molecular structure and crystal packing of 1a
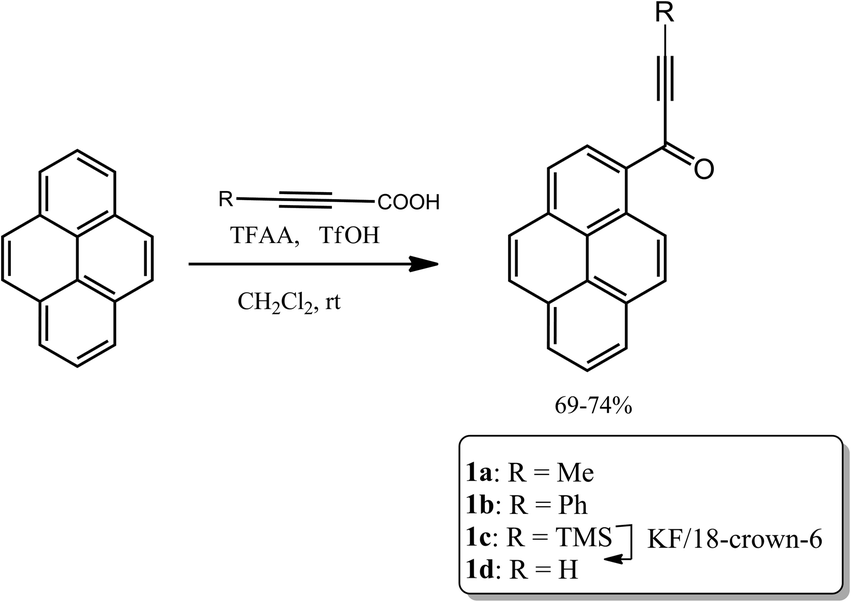
Crystals of 1a that were suitable for X-ray diffraction study were obtained by slow diffusion of n-heptane into a solution of this compound in chloroform. Under these conditions 1a crystallized in the centrosymmetric P21/c space group in a monoclinic system, with one independent molecule in the crystallographic asymmetric unit located in general position. The molecular structure of 1a is presented in Fig. 1.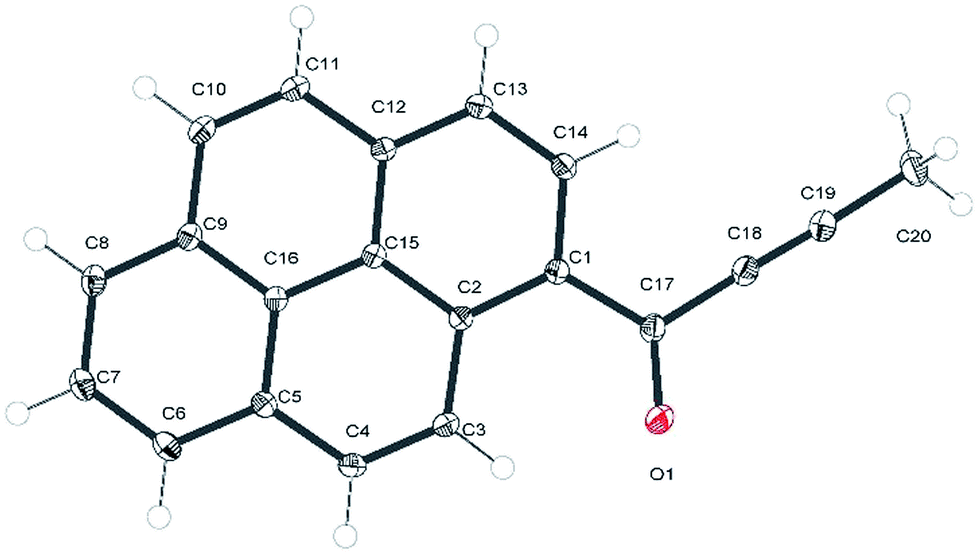 | ||
| Fig. 1 ORTEP representation of 1a with an atom numbering scheme. Atomic displacement parameters are drawn at 50% probability level. | ||
In the experimental structure of 1a the pyrenyl moiety is not ideally planar, but slightly bent along its longer (C7 → C14) axis. The angle between the plane of ring C5, C6, C7, C8, C9, and C16 (ring 1) and that of ring C1, C2, C12, C13, C14, C15 (ring 2) is 3.7(3) degrees. The propynoyl substituent is twisted out of the plane of adjacent ring 2, as indicated by the C2–C1–C17–C18 and C14–C1–C17–O1 torsion angles which were significantly different from 180° (172.37(7) degrees and C14 C1 C17 O1 168.68(9) degrees, respectively). The angle between the plane of ring 2 and the plane of the carbonyl moiety was 9.9(5) degrees. This conformation enables the formation of two weak intermolecular C–H⋯O hydrogen bonds in the crystal lattice: C4–H4⋯O1_i O1–H4 where the distance is 2.552(3) Å and C20–H20A⋯O1_ii O1–H20A where the distance is 2.579(3) Å (where (i) denotes the following symmetry transformation: 1 − x, 2 − y, 1 − z and (ii) denotes the symmetry transformation: x, 1 1/2 − y, 1/2 + z) (Fig. 2).
 | ||
| Fig. 2 C–H⋯O hydrogen bonds in a crystal structure of 1a. The atoms involved in the hydrogen bonds are presented with thermal ellipsoids at 50%, the remaining atoms are represented in grey for clarity. The hydrogen bonds are highlighted in cyan. The H⋯A distances are reported in Å. | ||
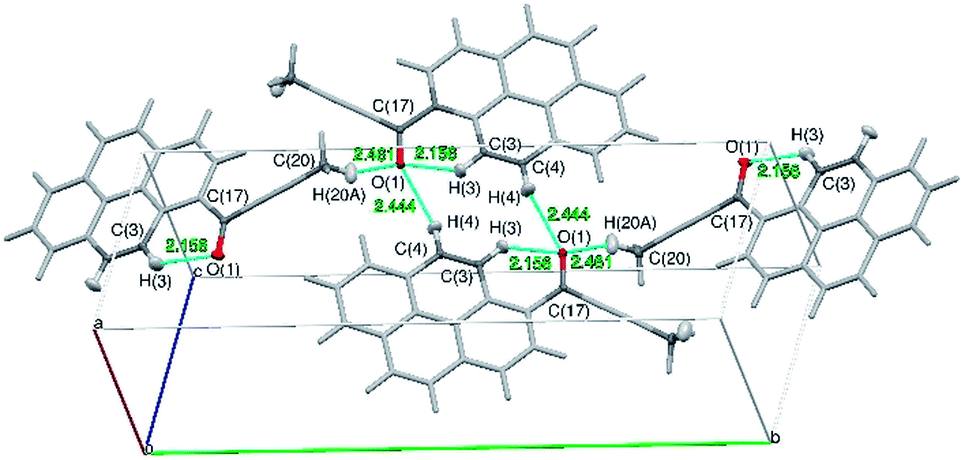
The tilt of the carbonyl group out of the ring 2 plane does not prevent the formation of a weak intramolecular C3–H3⋯O1 hydrogen bond, with an O1⋯H3 distance of only 2.156(1) Å. Viewed along the crystallographic [001] direction, the crystal packing of 1a shows a characteristic herringbone motif (Fig. 3a) with distinct layers of molecules stacked along the [100] direction. Within the layers the carbonyl moieties are oriented almost parallel to the [001] direction; in consecutive layers the carbonyl groups point in opposite directions (i.e. the carbonyl moieties in one layer are almost parallel to [001] direction, while the carbonyl moieties from the next layer are almost antiparallel to this direction). The angle between the C17–O1 carbonyl bond and the crystallographic [001] direction is 25.8(5) degrees (for a molecule in the alternative layer this would be 205.8(5) degrees).
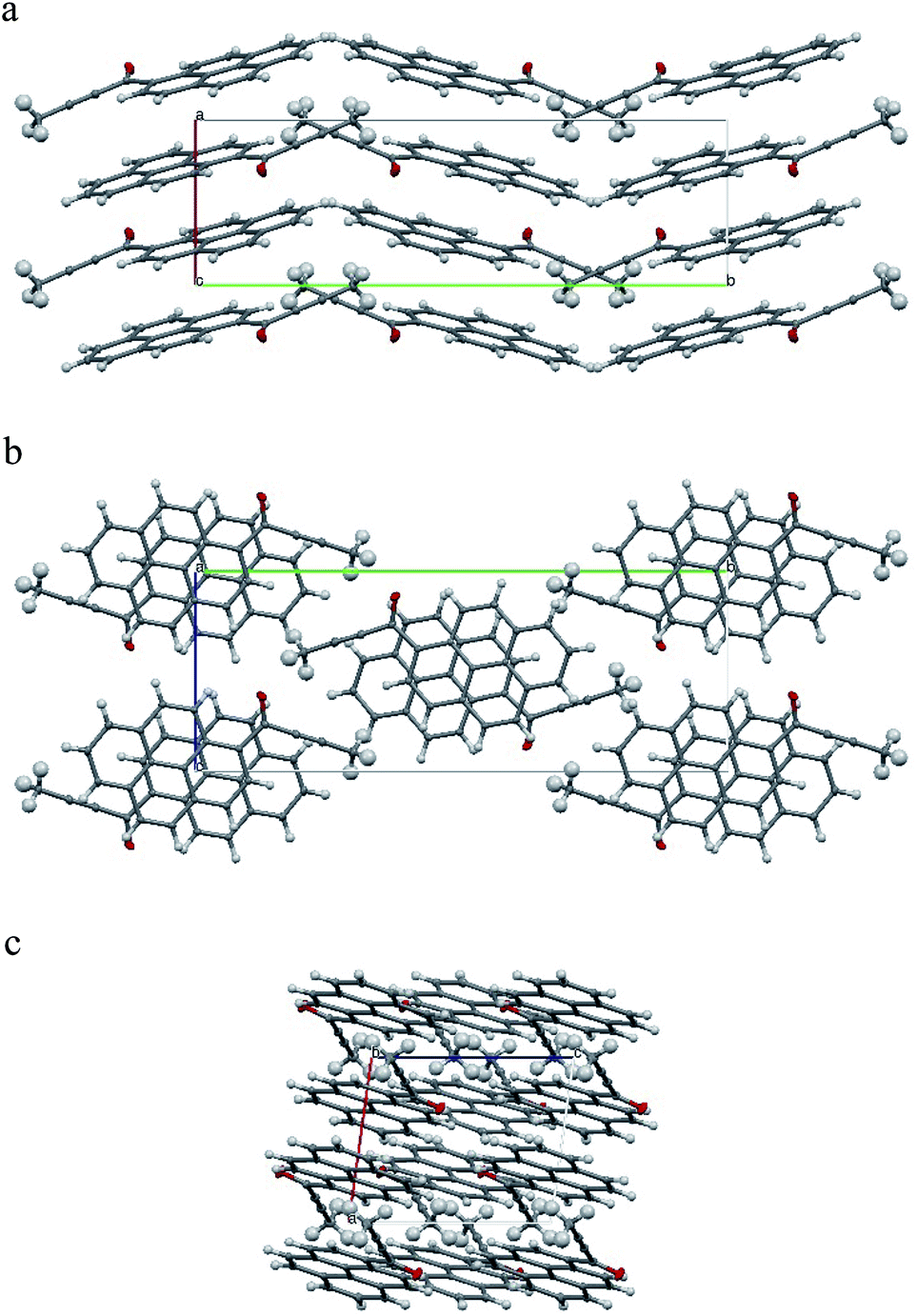 | ||
| Fig. 3 Crystal packing of 1a. (a) view along the [001] direction, (b) view along the [100] direction, (c) view along the [010] direction. Crystal axes denoted as follows: a [100] as red, b [010] as green and c [001] as blue. | ||
The crystal structure of 1a can be considered as composed of layers of molecules with parallel dipole and transition moments; with the directions of these moments alternating between layers. Each layer can be further viewed as composed of strands of molecules, packed along the [001] direction.
Molecules from consecutive layers along the [100] direction (Fig. 3b), related by crystallographic centres of inversion, are also involved in π-stacking interactions, thus building columns of symmetry-related molecules along [100] (Fig. 3a). Due to the ‘bend’ in the pyrene moiety, the closest contacts of an independent molecule (I) and the symmetry-related molecule II (above) are different from the closest contacts of an independent molecule (I) and the symmetry-related molecule III (below) (Fig. 4). For the former the closest contacts are C2_I–C12_II (3.377(3) Å) and C14_I–C16_II (3.419(3) Å), while for the latter the closest contacts are C1_I–C9_III (3.361(3) Å) and C15_I–C15_III (3.433(3) Å).
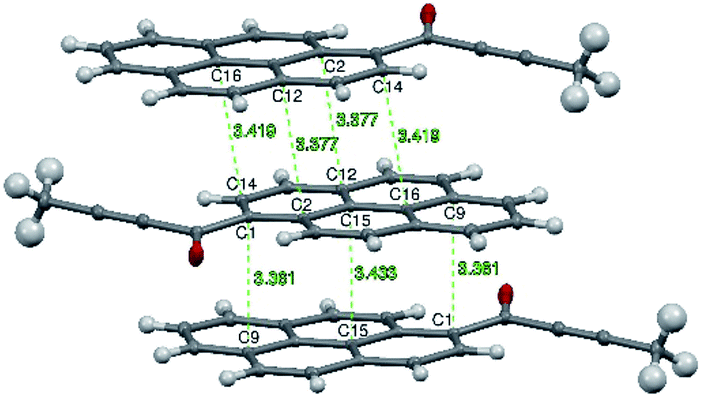 | ||
| Fig. 4 Intermolecular contacts in layers of 1a molecules along the [100] direction. The C⋯C distances are highlighted in green and reported in Å. Atomic displacement parameters are reported at 50% probability level. | ||
Interlayer interactions are stabilised by a weak C–H⋯O hydrogen bond: C4–H4⋯O1_i, while the separate layers are stabilised by a weak C–H⋯O hydrogen bond: C20–H20A⋯O1.
Because the scattering power of the 1a crystals was good, aspherical atomic scattering factors could be applied in structure refinement. Therefore, we could also perform a preliminary topological analysis of experimental charge density for this compound (see ESI†). It supports the structural analysis, thus confirming the presence and significance of all of the already mentioned C–H⋯O interactions. The strongest hydrogen bond that is present in the crystals of 1a is the intramolecular C3–H3⋯O1 bond, according to electron density and energy density criteria. A ring critical point was found within the C1–C2–C3–H3–O1–C17 ring, closed by this bond, thus confirming the significance of this interaction according to Koch and Popelier criteria.31 This analysis also demonstrated bond paths for the π-stacking interactions between symmetry-related molecules of 1a, which may support the ‘excimer’ hypothesis (vide infra). (Table 1)
| Interaction | ρ (rBCP) [e Å−3] | Lap (rBCP) [e Å−5] | G (rBCP) [H a0−3] | V (rBCP) [H a0−3] | H (rBCP) [H a0−3] | G (rBCP)/ρ (rBCP) [H e−1] | H (rBCP)/ρ (rBCP) [H e−1] | |V (rBCP)|/G (rBCP) |
|---|---|---|---|---|---|---|---|---|
| O1⋯H3 | 0.110 (5) | 2.076 (4) | 0.017 | −0.013 | 0.004 | 1.043 | 0.245 | 0.765 |
| H4⋯i_O1 | 0.056 (5) | 0.684 (3) | 0.006 | −0.004 | 0.001 | 0.723 | 0.121 | 0.667 |
| H20A⋯ii_O1 | 0.045 (4) | 0.745 (3) | 0.006 | −0.004 | 0.002 | 0.900 | 0.300 | 0.667 |
| C2⋯iii_C12 | 0.031 (1) | 0.295 (1) | 0.002 | −0.002 | 0.001 | 0.435 | 0.218 | 1.000 |
| C14⋯iii_C16 | 0.029 (1) | 0.272 (1) | 0.002 | −0.002 | 0.001 | 0.465 | 0.233 | 1.000 |
| C1⋯iv_C9 | 0.036(2) | 0.314 (1) | 0.003 | −0.002 | 0.001 | 0.562 | 0.187 | 0.667 |
| C15⋯iv_C15 | 0.021 (2) | 0.303 (1) | 0.002 | −0.001 | 0.001 | 0.643 | 0.321 | 0.500 |
The molecule of 1a in the crystal is strongly polarized with a negative charge of over 0.5e localised on the butynoyl group and an equivalent positive charge residing on the pyrene moiety. This results in a significant molecular dipole moment, oriented in between the C1–C17 and C17–O1 bond axes and coplanar with the pyrene moiety. According to the DFT B3PW91 calculations, an isolated molecule of 1a has the dipole moment of 3.8 D, while the dipole moment obtained from the experimental charge density model is almost 4 times larger (15.8 D). It must be stressed that the absolute value of the molecular dipole moment in the crystal cannot be reliably derived from the current experimental charge density model, and that the value only indicates a tendency for increased polarisation of the molecule in the crystalline environment. Quantum chemical calculations in the periodic lattice performed at the B3LYP level of theory with a 6-31+g(d) basis set yielded an optimised geometry that was identical to the geometry from X-ray diffraction within an experimental error. These calculations confirm enhancement of the molecular dipole moment in the crystal lattice. The resulting dipole moment has a direction that is in agreement with the results of experimental charge density analysis and the results of theoretical calculations for an isolated molecule and a value of 4.7 D. The dipole moment vector is almost perpendicular to the [100] crystallographic direction (∼86°) and coplanar with the pyrene moiety.
Photophysical properties of 1a–d and AcPyr in solution
The electronic absorption and emission spectra of 1a–d and AcPyr in chloroform (c = 10−6 M) are shown in Fig. 5 and the spectral data are collected in Table 2. At such a low concentration the formation of aggregates and excimers is highly unlikely and the observed emission can be ascribed to fluorophore monomers.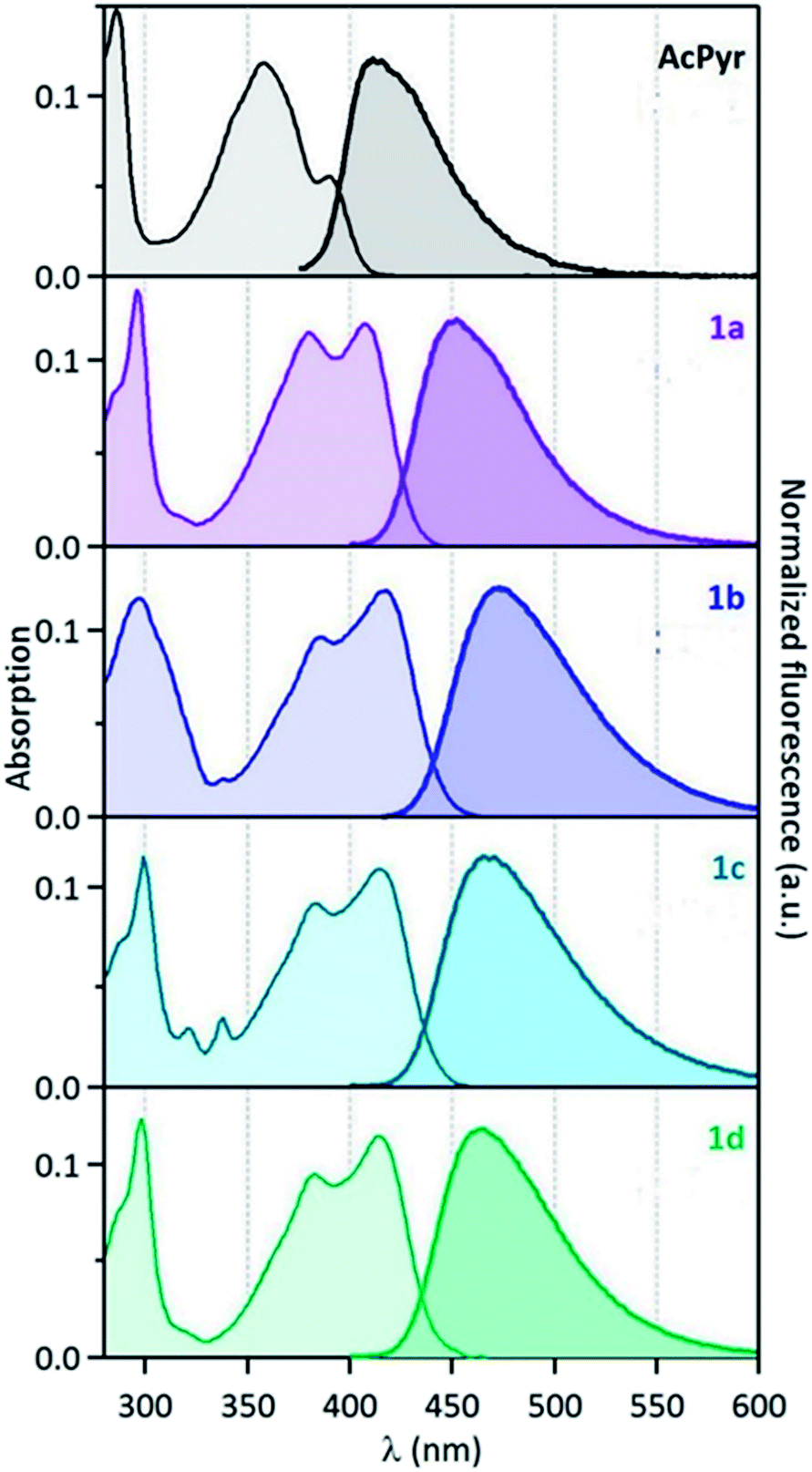 | ||
| Fig. 5 Normalised absorption (left) and fluorescence (right) spectra of compounds 1a–d and AcPyr in CHCl3 (λexc = 390 nm for 1a–d and 330 nm for AcPyr). | ||
| 1a | 1b | 1c | 1d | AcPyr | |
|---|---|---|---|---|---|
| λabs (nm) (CHCl3) | 408 | 418 | 416 | 415 | 391 |
| εmax (M−1 cm) (CHCl3) | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
45500 |
18800 |
24800 |
15400 |
| λem (nm) (CHCl3) | 449 | 471 | 462 | 460 | 409 |
| ΦF (CHCl3) | 0.02 | 0.06 | 0.05 | 0.07 | <0.005 |
| λem (nm) (solid) | 569 | 530 | 550 | 565 | 486 |
| ΦF (solid) | 0.13 | 0.04 | 0.12 | 0.07 | — |
| τ1 (ns)/contribution (CHCl3) | 0.11/0.67 0.29/0.29; 0.73/0.04 | 0.24/0.44; 0.77/0.56 | 0.20/0.650.60/0.35 | 0.24/0.250.68/0.75 | 0.06/0.890.27/0.11 |
The spectra reveal significant (up to ∼60 nm) bathochromic shifts of the bands of 1a–d in comparison to those of AcPyr. This suggests that the alkynoyl substituents are more efficiently conjugated with the pyrenyl moiety than the acetyl group. Furthermore, the absorption bands of 1a–d enter the visible region and these compounds can be excited with violet light. All of the investigated ketones are practically non-fluorescent in a nonpolar solvent, hexane (see ESI†). In a medium polarity solvent, chloroform, compounds 1a–d showed fluorescence at 449–471 nm with quantum yields in the range of 0.02–0.07, whereas AcPyr emitted weakly at 409 nm (quantum yield was lower than 0.005). In a more polar aprotic solvent, DMSO, all 1a–d were emissive, whereas in a polar but hydroxylic solvent, methanol, only 1a was a strong emitter. Surprisingly, 1a–d were stronger emitters in chloroform than in methanol, whereas the opposite effect was observed for AcPyr. Time-resolved fluorescence investigations of 1a–d and AcPyr were performed in chloroform solutions and revealed multiexponential decays in all cases (Fig. 6a, Table 2). A fast decay was observed for AcPyr, intermediate behavior was observed for 1a and 1c, whereas much slower decays were recorded for 1b and 1d. Three sets of decay time constants could be distinguished: a fast contribution (τ1 < 0.15 ns), an intermediate contribution (τ2 = 0.2–0.3 ns), and a slow contribution (τ3 > 0.5 ns).
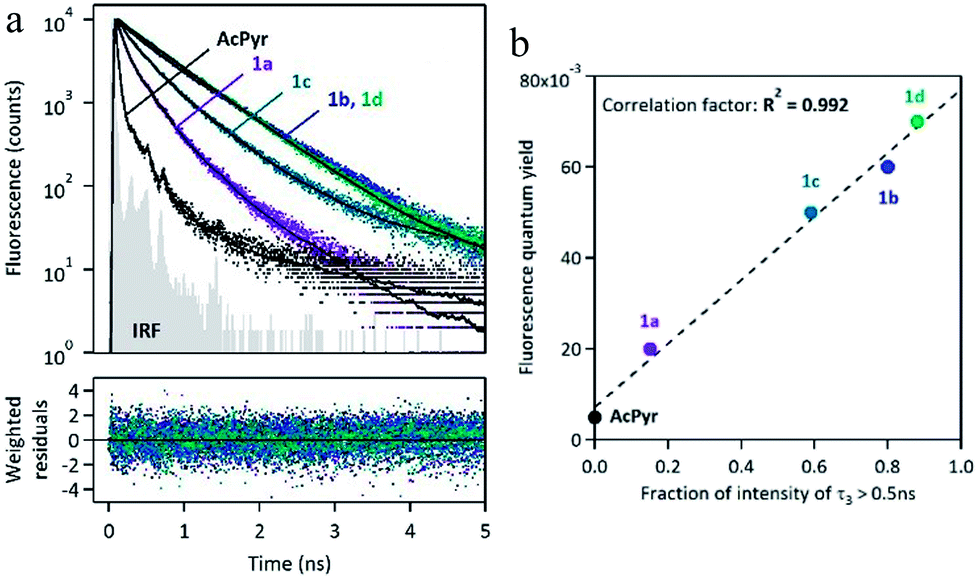 | ||
| Fig. 6 (a) Fluorescence decay curves of 1a–d and AcPyr recorded in CHCl3, with multiexponential fitting and weighted residuals (IRF = instrumental response function). (b) Correlation plot between the fluorescence quantum yield measured in CHCl3 and the fraction of intensity corresponding to the slowest contribution (typically with a decay time constant >0.5 ns). | ||
These three contributions may be related to three populations of conformers having different relaxation times and different emission efficiencies. The fraction of intensity related to the slowest component τ3 is very much variable depending on the compound. It is absent for AcPyr, represents a small proportion for 1a (ϕ3 = 0.15) and its contribution becomes predominant for compounds 1b (ϕ3 = 0.80), 1c (ϕ3 = 0.59), and 1d (ϕ3 = 0.88). This fraction of intensity is well-correlated to the overall fluorescence quantum yields of compounds AcPyr and 1a–d (Fig. 6b). Therefore, we could conclude that the slowest decay time corresponds to a conformer which is much more fluorescent than the others. Consequently, the fluorescence quantum yields measured in CHCl3 reflect the various proportions of the different conformers, which are variable from one compound to another.
Solid-state fluorescence of 1a–d
In contrast to AcPyr, which is almost non-emissive in the solid state (it exhibits extremely weak fluorescence centred at 486 nm), ketones 1a–d are moderate solid-state emitters (Fig. 7 and 8 and Table 2). The solid state fluorescence of these compounds is centred at 530–567 nm, i.e. ∼100 nm red-shifted compared to solution emission. This large shift suggests that the emissive state may be rather different to that of the molecular monomer in solution. The fluorescence quantum yields are in the range of 0.04 (1b)–0.13 (1a). Compounds 1b and 1d did not show a noticeable increase in solid-state fluorescence quantum yields as compared to the CHCl3 solution values, whereas 1a showed a 6-fold and 1c a 2.5-fold increase of the quantum yield. Moreover, solid-state excitation spectra were also very much red-shifted as compared to the CHCl3 solution, with λmax of the first band in the 430–470 nm range, corresponding to red-shift values from 20 nm (1a) up to 55 nm (1d). | ||
| Fig. 7 Photographs of 1a–d and AcPyr in the solid state under visible light (top row) und under 254 nm UV light (bottom row) illumination. | ||
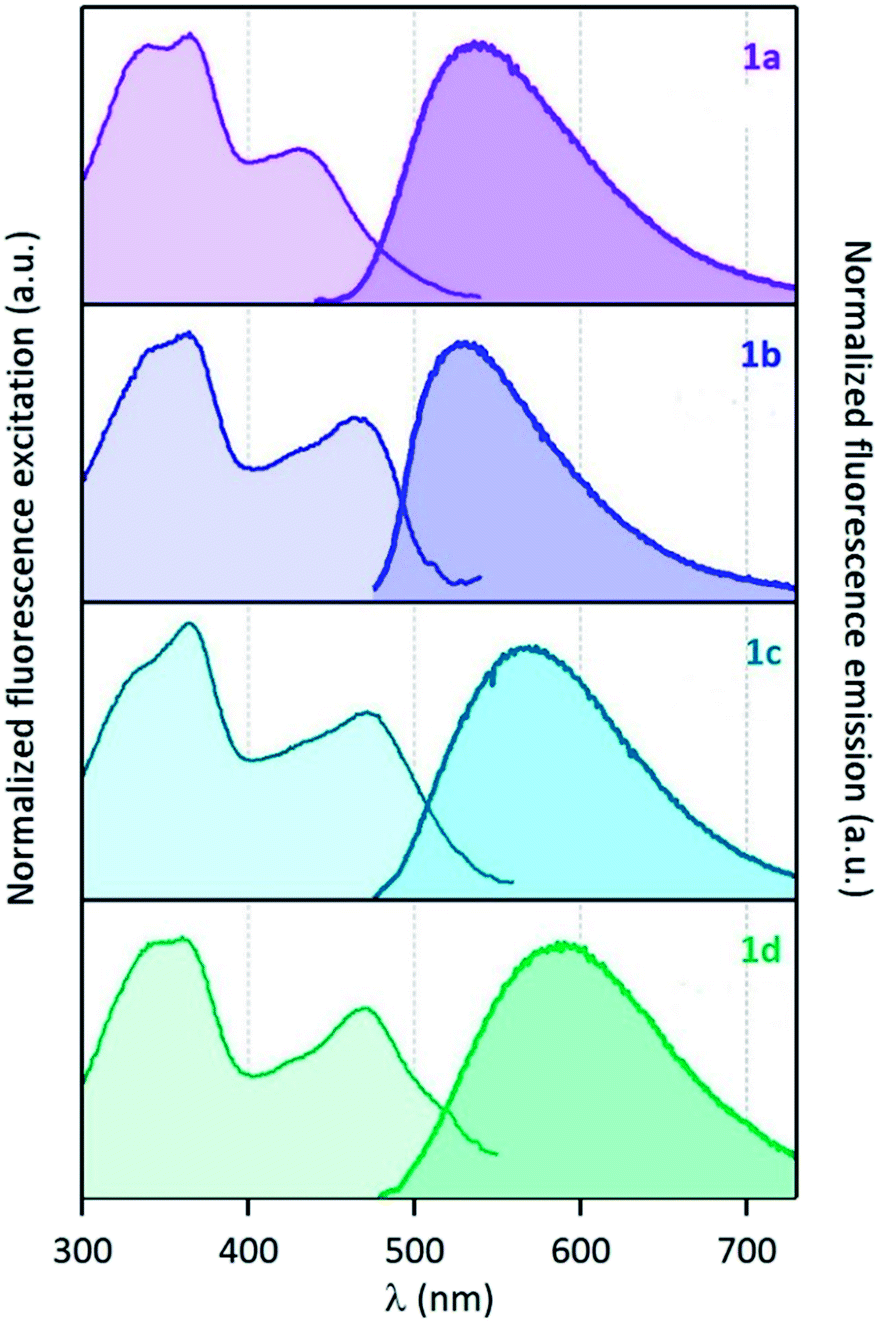 | ||
| Fig. 8 (Left) normalized excitation and (right) emission spectra of compounds 1a–d recorded in the solid state (powder inserted in an integration sphere). λexcit = 430 nm for 1a and 460 nm for 1b–d. | ||
Fluorescence time-resolved experiments of 1a were also performed in the solid state under ambient atmosphere. Fluorescence decay curves monitored at three different emission wavelengths (λem = 540, 590 and 640 nm) are shown in Fig. 9.
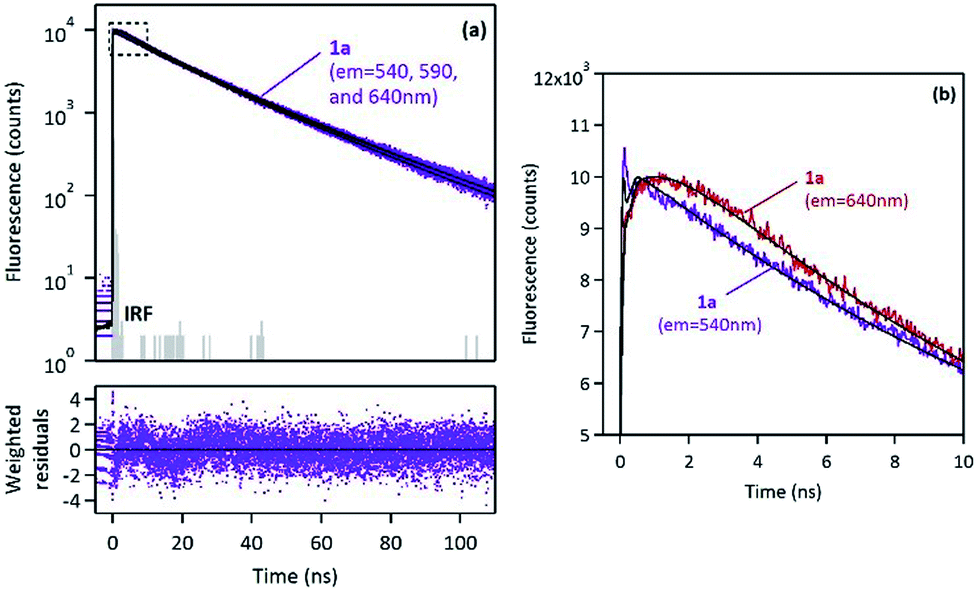 | ||
| Fig. 9 (a) Fluorescence decay curves of 1a in the solid state, monitored at three different emission wavelengths (λem = 540, 590 and 640 nm), with multiexponential fitting and weighted residuals. (b) Zoomed region at short times, where excimer-type rise-time is clearly observed at λem = 640 nm, but absent at λem = 540 nm. | ||
Global analysis was successfully applied to the three decays with multiexponential fitting and revealed three common components: two long decay times, τ1 = 13.2 ns and τ2 = 27.4 ns (which represented more than 99% of the fraction of intensity) and a short component, τ3 ∼ 0.6–1.2 ns, which appeared either as a decay time (for λem = 540 nm) or as a rise-time (for λem = 590 and 640 nm), as shown in Fig. 9. Such a short decay time in the blue-edge of the spectrum corresponding to a short rise-time in the red part of the spectrum is a typical signature of excimer formation which occurs within the crystal with a fast kinetic rate. This mechanism is rather consistent with the face-to-face orientation of the pyrene moieties in the crystal lattice, as revealed by X-ray diffraction (vide supra).
It is generally believed that such an aggregation leads to non-emissive species (H-aggregates). However, some examples of emissive H-aggregates have been reported.32–34 In such aggregates, emission may arise from restriction of intramolecular rotation (RIR), blocking of nonradiative decay channels and from the formation of emissive solid-state excimers (meaning not only “excited dimers” but also “excited oligomers”). Consequently, 1a–d emitted in the solid-state as monomers (a green emission), but they also interacted very quickly in the excited state and led to strongly emissive excimer species (a red emission), and then following a very slow decay rate. This situation is different from the one observed in solution, where only fast monomer decays were observed.
It should be emphasized that solid-state fluorescence, which is essential for various industrial applications, is still a relatively rare phenomenon because of the ubiquitous aggregation-caused quenching (ACQ) effect. Solid-state fluorophores exhibiting the aggregation-induced emission enhancement (AIEE) effect were discovered only very recently.35–37 Moreover, such fluorophores bearing a flat π-conjugated pyrene moiety are relatively rare.38–40 Strong yellow solid-state emission of 1a–d, with long fluorescence decays, makes them promising candidates for the design of luminescent materials and devices.
Comparative DFT and TD DFTstudy of 1d and AcPyr
To gain deeper insight into the influence of an acetylenic bond on the electronic structure of pyrene carbonyl chromophore DFT and TD DFT calculations were performed on the simplest ynone (1d) and AcPyr. It has to be mentioned that a DFT study of the latter molecule was recently published by Konishi et al.15 However, in the present work we focused our interest on a comparison of both compounds at the same calculation level. The B3PW91 functional with a 6-311+g(d) basis set was used since it provided satisfying results for structurally close alkynoylferrocenes.41First we scanned the potential energy vs. the dihedral angle between the pyrenyl moiety and the CO group (approximated by the C2–C1–C17–O1 angle), for 1d and AcPyr (Fig. 10).
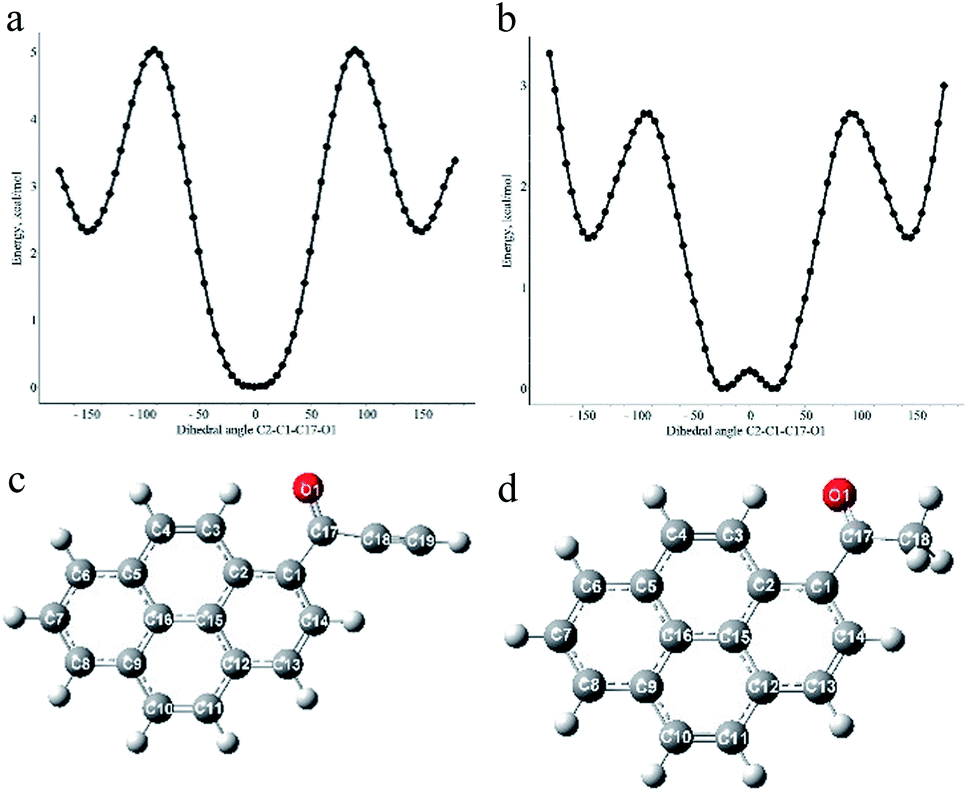 | ||
| Fig. 10 Plots of calculated energy vs. C2–C1–C17–O1 dihedral angle for 1d (a) and AcPyr (b) and optimized geometries of the most stable conformations of these compounds (c) and (d). | ||
The most stable geometry of 1d is planar with the carbonyl oxygen interacting with C3–H (Fig. 10a and c). The same conformation is present in the crystals of 1a (vide supra), a small twist of the CO group being attributable to the packing effects.
It is more stable by 2.34 kcal mol−1 than the conformation with the carbonyl oxygen directed towards C14–H and having the C2–C1–C17–O1 dihedral angle 150°. This means that the equilibrium amount of the latter conformer at room temperature is <3%. The calculated barrier to rotation of the carbonyl group is 5.03 kcal mol−1. On the other hand, the most stable conformation of AcPyr (Fig. 10b and d) is nonplanar with a C2–C1–C17–O1 dihedral angle 25° and with the carbonyl oxygen also interacting with C3–H (the corresponding planar conformation is slightly less stable by 0.18 kcal mol−1). However, in this case the conformation with the dihedral CO/pyrene angle equal to 145° is less stable only by 1.49 kcal mol−1, which means that the equilibrium amount of this conformation at room temperature may reach 8–10%. The energy barrier to rotation of the acetyl group was found to be equal to 2.72 kcal mol−1. It is also worth noting that the pyrenyl-CO bond in 1d is significantly shorter than the analogous bond in AcPyr (1.483 and 1.495 Å, respectively). This reveals a more efficient pyrenyl-CO conjugation in 1d, which is in line with the higher energy barrier for rotation around this bond in this compound than in AcPyr.
The calculated molecular orbitals of 1d and AcPyr are shown in Fig. 11
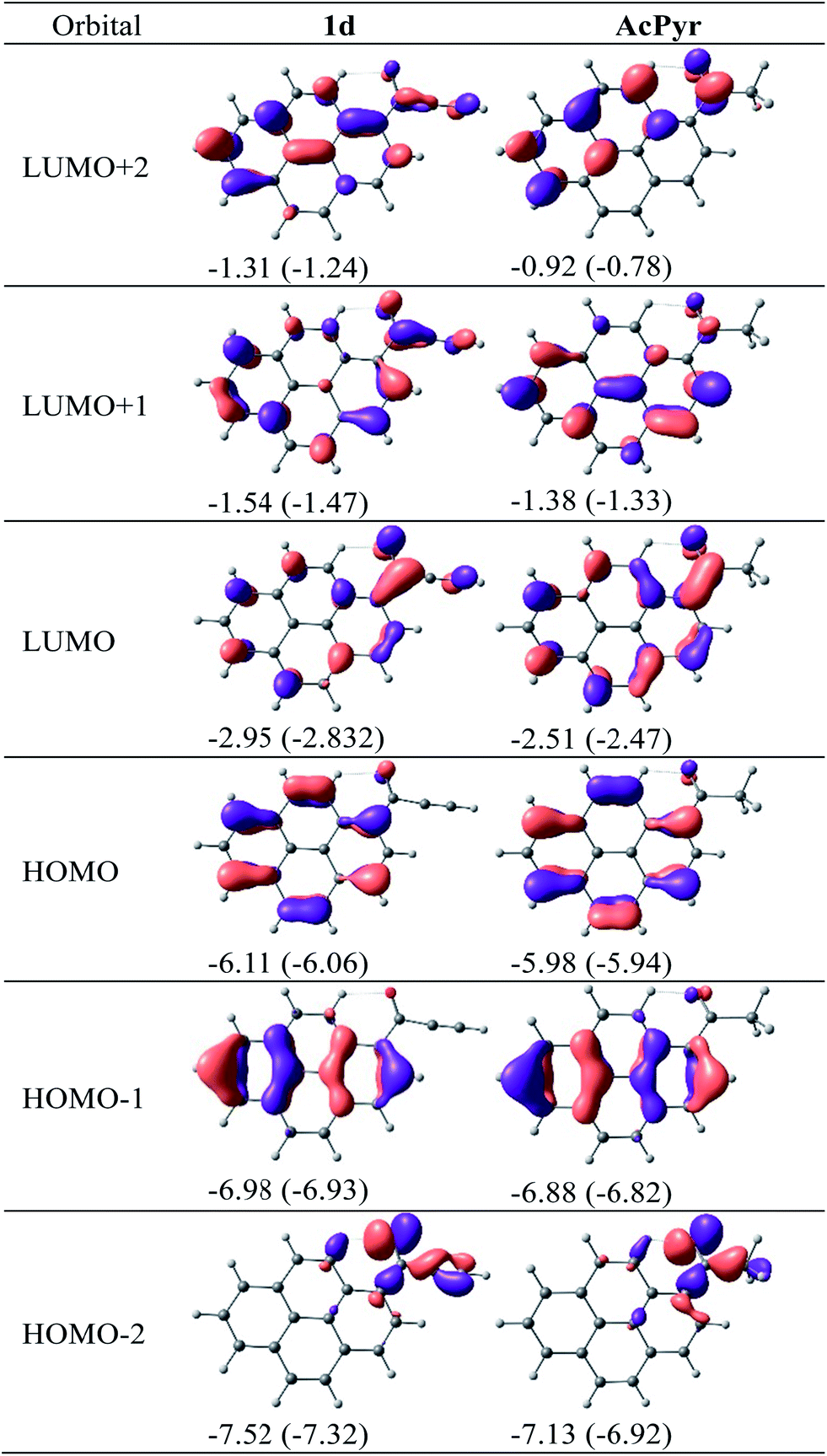 | ||
| Fig. 11 DFT-calculated molecular orbitals for 1d and AcPyr. Orbital energies in eV in vacuum and (in parentheses) in CHCl3. | ||
The HOMO-1 and HOMO of 1d and AcPyr differ only slightly in energy (∼0.1 eV), presumably because in these orbitals the electron density is localized mostly at the pyrene moiety. In contrast, the HOMO-2 orbitals of these compounds are localized on the acyl groups, which results in a stronger (0.4 eV) stabilization of this orbital in 1d. A significant difference in localization of electron density is observed for unoccupied LUMO, LUMO + 1 and LUMO + 2 orbitals. In the case of AcPyr these orbitals are localized on the pyrenyl moiety and the carbonyl group, whereas delocalization is observed on the ethynyl group in the case of 1d. The strongest stabilization (∼0.4–0.5 eV) is observed for the LUMO and LUMO + 2 orbital of 1d, in comparison to the same orbital in AcPyr.
We have also calculated the electronic absorption spectra of 1d and AcPyr for isolated molecules and for a chloroform solution using time-dependent DFT and a polarizable continuum model (PCM) using integral equation formalism variant (IEFPCM). The data are gathered in Table 3.
| Compound | λmax/ nm (eV) | f | Main contribution(s) |
|---|---|---|---|
| (a) Isolated molecules | |||
| 1d | 407.4 (3.04) | 0.3985 | H → L (0.95) |
| 356.1 (3.48) | 0.0527 | H-1 → L (0.79) | |
| 292.1 (4.24) | 0.1297 | H → L + 1 (0.71) | |
| 261.9 (4.73) | 0.1231 | H-4 → L (0.79) | |
| AcPyr | 377.0 (3.29) | 0.3523 | H → L (0.92) |
| 283.5 (4.37) | 0.1909 | H → L + 1 (0.45) | |
| H-1 → L (0.19) | |||
| 247.6 (5.01) | 0.1743 | H-4 → L (0.62) | |
| (b) CHCl3 solution | |||
| 1d | 429.3 (2.89) | 0.5538 | H → L (0.97) |
| 362.3 (3.42) | 0.0774 | H-1 → L (0.85) | |
| 296.4 (4.18) | 0.2104 | H → L + 1 (0.70) | |
| 291.7 (4.25) | 0.0510 | H → L + 2 (0.73) | |
| 266.9 (4.64) | 0.1416 | H-4 → L (0.84) | |
| 249.9 (4.96) | 0.1003 | H-5 → L (0.89) | |
| AcPyr | 386.8 (3.21) | 0.4962 | H → L (0.95) |
| 298.7 (4.15) | 0.0525 | H-3 → L (0.64) | |
| H → L + 1 (0.22) | |||
| 287.0 (4.32) | 0.2435 | H → L + 1 (0.37) | |
| H-3 → L (0.32) | |||
The lowest energy band may practically be considered a pure HOMO–LUMO transition for both compounds. The calculated wavelengths of this band for the chloroform solution are in good agreement with the experimental values (429 vs. 415 nm for 1d and 387 vs. 391 nm for AcPyr). It is generally believed that face-to-face H-aggregates are non-emissive in terms of Kasha's theory of exciton coupling. However, some examples of emissive H-aggregates have been reported.32–34 In such aggregates, emission may arise from restriction of intramolecular rotation (RIR), blocking nonradiative decay channels or formation of emissive solid-state excimers (meaning not only “excited dimers” but also “excited oligomers”). A time-resolved fluorescence study clearly demonstrated that solid-state emission of 1a originated from dynamic excimers formed during and shortly after the laser pulse and emitting at longer wavelengths than monomeric molecules. Some contribution from the RIR effect is also possible since the intermolecular hydrogen bond network along with π-stacking may severely inhibit rotational motions in individual fluorophores in the crystals.
Conclusions
We elaborated an efficient method of Friedel–Crafts acylation of pyrene with conjugated alkynoic acids which led to a new class of pyrenyl fluorophores – 1-pyrenyl ynones. Comparison of the electronic absorption and fluorescence emission spectra of these compounds with those of the simplest saturated pyrenyl ketone, 1-acetylpyrene, revealed some of their advantages. The lowest energy absorption bands of 1-pyrenyl ynones were shifted bathochromically to the visible region, therefore these compounds can be conveniently excited with violet light. They exhibit more intense fluorescence than 1-acetylpyrene in a medium polarity environment. Their fluorescence maxima are shifted towards lower energies by 50–60 nm and their fluorescence lifetimes are substantially longer than those of 1-acetylpyrene. In contrast to the latter compound, they exhibit strong fluorescence in the solid state, which is associated with long decay times and which can be attributed to an efficient formation of excimer species. Finally, the simplest ynone, 1-(pyren-1-yl)prop-2-yn-1-one, may be used in azide-alkyne click chemistry for introduction of a fluorescent pyrenyl tag to diverse molecular structures (biomolecules, polymers, etc.) This possibility is currently being studied in our laboratory and the results will be published in due time.Acknowledgements
Financial support from the National Science Centre (Grant Harmonia UMO-2012/04/M/ST5/00712) is gratefully acknowledged. This research was also supported in part by the PL-Grid Infrastructure.Notes and references
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC.
- A. M. Breul, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 5366–5407 RSC.
- G. Drummen, Molecules, 2012, 17, 14067–14090 CrossRef CAS PubMed.
- G. Bains, A. B. Patel and V. Narayanaswami, Molecules, 2011, 16, 7909–7935 CrossRef CAS PubMed.
- R. Métivier, I. Leray, M. Roy-Auberger, N. Zanier-Szydlowski and B. Valeur, New J. Chem., 2002, 26, 411–415 RSC.
- R. Métivier, I. Leray, J.-P. Lefevre, M. Roy-Auberger, N. Zanier-Szydlowski and B. Valeur, Phys. Chem. Chem. Phys., 2003, 5, 758–766 RSC.
- A. Bencini and V. Lippolis, Coord. Chem. Rev., 2012, 256, 149–169 CrossRef CAS PubMed.
- J. Xie, M. Ménand, S. Maisonneuve and R. Métivier, J. Org. Chem., 2007, 72, 5980–5985 CrossRef CAS PubMed.
- L. Zöphel, V. Enkelmann and K. Müllen, Org. Lett., 2013, 15, 804–807 CrossRef PubMed.
- X. Feng, J.-Y. Hu, F. Iwanaga, N. Seto, C. Redshaw, M. R. J. Elsegood and T. Yamato, Org. Lett., 2013, 15, 1318–1321 CrossRef CAS PubMed.
- Y. Niko, S. Kawauchi, S. Otsu, K. Tokumaru and G.-i. Konishi, J. Org. Chem., 2013, 78, 3196–3207 CrossRef CAS PubMed.
- M. Ottonelli, M. Piccardo, D. Duce, S. Thea and G. Dellepiane, J. Phys. Chem. A, 2012, 116, 611–630 CrossRef CAS PubMed.
- C. X. Yao, H. B. Kraatz and R. P. Steer, Photochem. Photobiol. Sci., 2005, 4, 191–199 CAS.
- Y. Niko, Y. Hiroshige, S. Kawauchi and G.-i. Konishi, Tetrahedron, 2012, 68, 6177–6185 CrossRef CAS PubMed.
- Y. Niko, Y. Hiroshige, S. Kawauchi and G. I. Konishi, J. Org. Chem., 2012, 77, 3986–3996 CrossRef CAS PubMed.
- Y. Niko, S. Kawauchi and G. I. Konishi, Tetrahedron Lett., 2011, 52, 4843–4847 CrossRef CAS PubMed.
- A. Jana, S. Atta, S. K. Sarkar and N. D. P. Singh, Tetrahedron, 2010, 66, 9798–9807 CrossRef CAS PubMed.
- K. Szczubiałka, Ł. Moczek, A. Goliszek, M. Nowakowska, A. Kotzev and A. Laschewsky, J. Fluorine Chem., 2005, 126, 1409–1418 CrossRef PubMed.
- L. Bucsiova, P. Hrdlovič and Š. Chmela, J. Photochem. Photobiol., A, 2001, 143, 59–68 CrossRef CAS.
- Y. Niko and G.-i. Konishi, J. Synth. Org. Chem Jpn., 2012, 70, 918–927 CrossRef CAS.
- D. A. Fleming, C. J. Thode and M. E. Williams, Chem. Mater., 2006, 18, 2327–2334 CrossRef CAS.
- S. P. Sau and P. J. Hrdlicka, J. Org. Chem., 2011, 77, 5–16 CrossRef PubMed.
- D. Plażuk and J. Zakrzewski, J. Organomet. Chem., 2009, 694, 1802–1806 CrossRef PubMed.
- B. Willy and T. J. J. Müller, ARKIVOC, 2008, i, 195–208 CrossRef.
- M. Navidi and B. Movassagh, Monatsh. Chem., 2013, 144, 1363–1367 CrossRef.
- W. Sun, Y. Wang, X. Wu and X. Yao, Green Chem., 2013, 15, 2356–2360 RSC.
- B. Huang, L. Yin and M. Cai, New J. Chem., 2013, 37, 3137–3144 RSC.
- M. Navidi, B. Movassagh and S. Rayati, Appl. Catal., A, 2013, 452, 24–28 CrossRef CAS PubMed.
- N. M. Carballeira, Chem. Phys. Lipids, 2013, 172–173, 58–66 CrossRef CAS PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- V. Karunakaran, D. D. Prabhu and S. Das, J. Phys. Chem. C, 2013, 117, 9404–9415 CAS.
- U. Rösch, S. Yao, R. Wortmann and F. Würthner, Angew. Chem., Int. Ed., 2006, 45, 7026–7030 CrossRef PubMed.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS.
- S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS.
- M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Q. Feng, M. Wang, B. Dong, C. Xu, J. Zhao and H. Zhang, CrystEngComm, 2013, 15, 3623–3629 RSC.
- P. Kotchapradist, N. Prachumrak, R. Tarsang, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, J. Mater. Chem. C, 2013, 1, 4916–4924 RSC.
- Y. Li, D. Wang, L. Wang, Z. Li, Q. Cui, H. Zhang and H. Yang, J. Lumin., 2012, 132, 1010–1014 CrossRef CAS PubMed.
- D. Plażuk, J. Zakrzewski, K. Nakatani, A. Makal, K. Woźniak and S. Domagala, RSC Adv., 2012, 2, 3512–3524 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses of 1a–d. Electronic absorption and emission spectra of 1a–d in various solvents. Details of X-ray diffraction and photophysical studies. CCDC 996918. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra03961k |
| This journal is © The Royal Society of Chemistry 2014 |