
Novel transmetalation reaction for electrolyte synthesis for rechargeable magnesium batteries†
Abstract
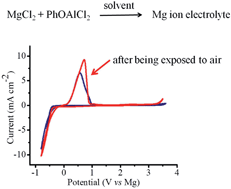
A simple strategy for the synthesis of electrolyte solutions comprised of binuclear magnesium aluminate complexes without the need for organomagnesium compounds is established. The as-prepared phenolate based electrolyte exhibits an anodic stability of up to 3.4 V, good ionic conductivity and air-stability.
 Please wait while we load your content...
Please wait while we load your content...

