
High-molecular-weight polar acrylate block copolymers as high-performance dismantlable adhesive materials in response to photoirradiation and postbaking
Abstract
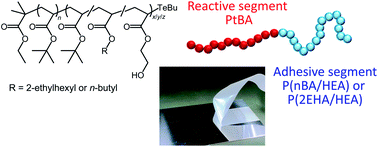
We synthesized high-molecular-weight acrylate block copolymers as high-performance dismantlable adhesives consisting of a poly(tert-butyl acrylate) (PtBA) sequence as the reactive segment and a random copolymer sequence of n-butyl acrylate (nBA) or 2-ethylhexyl acrylate (2EHA) with 2-hydroxyethyl acrylate (HEA) as the adhesive segment, using an organotellurium-mediated living radical polymerization (TERP). The adhesion strength of PtBA/P2EHA and PtBA/PnBA block copolymers containing polar HEA repeating units in their soft segments was sufficiently high for use as a pressure-sensitive adhesive. A quick change in the adhesion properties was observed in response to the dual external stimuli of photoirradiation and postbaking during the dismantling process. We discuss the adhesion strength and failure mode as a function of the HEA content, the sequence structure of the copolymers, and the external stimulus conditions.
 Please wait while we load your content...
Please wait while we load your content...

