
Is surface patch binding between proteins symmetric about isoelectric pH?†
Abstract
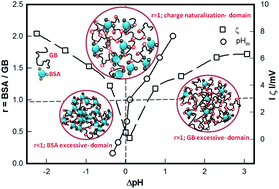
Surface selective patch binding (SPB) interaction occurring between two protein molecules, bovine serum albumin (BSA) and gelatin B (GB), both having same isoelectric pH (pI ≈ 5) and identical pH-zeta potential profile, was systematically examined. BSA : GB mixing ratio r was varied in the range 0.16–2.00 and ionic strength was varied in the range 0–10 mM, which yielded optimum binding ratio r = 1. The binding profiles produced asymmetric bell-like curves with clearly identifiable pairs of transition pHs: onset of intermolecular interaction, formation of soluble complexes and coalescence of the soluble complexes occurring at pHc1,2, pHφ1,2 and pHm respectively. Since pHm could be approached from either lower or higher side of pI, these profiles yielded pairs of pHc and pHφ values. In fact, we found (pHc2 − pI) > (pHc1 − pI), which clearly indicated that initiation of intermolecular associative interaction was not symmetric about pI (pI = pHm for r ≤ 1, an observation not reported hitherto. Secondly, (pHφ2 − pI) ≈ (pHφ1 − pI) implied that the pH at which soluble complexes formed (pHφ) was always located symmetrically about pHm, irrespective of the binding ratio. Higher binding affinity determined from higher value of pHc2 was confirmed from size measurement results. The change in the turbidity maximum Δτ could be correlated as Δτ ∼ I1/2 implying electrostatic screening of SPB with increase in ionic strength (I). This interaction was modelled using a linear combination of attractive and repulsive electrostatic forces which revealed considerable screening of the interaction potential U, consistent with aforesaid experimental data; ΔU ∼ I1/2. Further, it is concluded that intermolecular binding in protein–polyampholyte systems is qualitatively different from that in protein–polyelectrolyte class.
 Please wait while we load your content...
Please wait while we load your content...

