
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced dielectric properties of sol–gel-BaTiO3/P(VDF-HFP) composite films without surface functionalization†
Claudia
Ehrhardt
*a,
Christian
Fettkenhauer
a,
Jens
Glenneberg
b,
Wolfram
Münchgesang
c,
Hartmut S.
Leipner
b,
Gerald
Wagner
b,
Martin
Diestelhorst
c,
Christoph
Pientschke
c,
Horst
Beige
c and
Stefan G.
Ebbinghaus
*a
aInstitute of Chemistry, Martin-Luther-University Halle-Wittenberg, Kurt-Mothes-Straße 2, D-06120 Halle (Saale), Germany. E-mail: claudia.ehrhardt@chemie.uni-halle.de; stefan.ebbinghaus@chemie.uni-halle.de; Fax: +49 345 5527028; Tel: +49 345 5525641 Tel: +49 345 5525870
bInterdisciplinary Centre of Materials Science, Martin-Luther-University Halle-Wittenberg, Heinrich-Damerow-Straße 4, D-06120 Halle (Saale), Germany
cInstitute of Physics, Martin-Luther-University Halle-Wittenberg, Von-Danckelmann-Platz 3, D-06120 Halle (Saale), Germany
First published on 5th August 2014
Abstract
Barium titanate/poly(vinylidenfluoride-co-hexafluoropropylene) (BTO/P(VDF-HFP)) nanocomposite films were prepared by spin coating and their dielectric properties were investigated. Ferroelectric BaTiO3 was prepared by a low temperature sol–gel soft-chemistry method leading to spherical nanoparticles with a high degree of surface hydroxyl groups. Phosphonic acids and/or additional dispersant are typically used to cover the surface of hydrophilic oxide particles in order to improve the compatibility between the particles and the hydrophobic polymer. The effect of such surface modifications was studied using 2,3,4,5,6-pentafluorobenzyl phosphonic acid and additionally the dispersant BYK-W 9010 as surfactants. Against expectation, the use of pure sol–gel barium titanate led to the lowest particle agglomeration in the composite suspension and the best film quality resulting in a large enhancement of the relative permittivity by a factor of 10 compared to pure P(VDF-HFP). For this simple biphasic system without any further components a very high relative permittivity εr of 57 was obtained at 1 kHz for a 50 vol% BaTiO3/P(VDF-HFP) composite. The εr values further increase at lower frequencies. These findings show that for the BTO nanoparticles, prepared by the sol–gel synthesis, no surface coating is required to obtain good dielectric properties, allowing for a considerable simplification of the preparation technique.
Introduction
Composite dielectrics are promising materials for energy storage systems because they combine the good processability and breakdown field strength of polymers with the high permittivity of oxides. In a biphasic composite the dielectric properties are affected by both constituents. Therefore, the challenge is to design a material with a high permittivity and dielectric field strength without a large increase in dielectric loss, with the aim to enhance the resulting energy density.1Nanocomposites are attractive because of their large interfacial interactions, which have a significant effect on the degree of polarisation and charge separation.2 The ferroelectric polarisation determines the materials properties at high frequencies and affects the polarisation mechanism, while for lower frequencies or DC conditions the interfacial space-charge effects are dominating.3–5 A uniform distribution of the filler particles in the matrix enhances the interaction between the components, resulting in an increased interfacial polarisation.6,7 This polarisation is due to the differences in the electrical properties (permittivity and conductivity), leading to charge accumulations at the boundaries to the less conducting regions.8 Different approaches have been used to enhance the composite permittivities e.g. by thermal treatment of the oxide particles, application of bimodal particle size distributions or increase of the filler volume fractions.9–14 On the other hand, the large contrast in permittivity or conductivity between filler and the polymer host often generates highly inhomogeneous electric fields, which reduce the effective breakdown strength of the composites by electrical defect centres.15–17 The use of nanoparticles decreases the occurrence of large local fields.18 Various investigations focused on the effect of oxide surface functionalization, e.g. with phosphonic acids, to prevent agglomeration and to improve the particles distribution and in turn the composite homogeneity with the aim to enhance the dielectric characteristics.11,19–23 It was shown that oxide particles coated with organo-phosphonates resulted in an improved dielectric breakdown resistance and energy storage density.24 On the other hand, such surfactants can also cause an increase of dielectric loss and leakage current due to residual free molecules.25 Therefore, residual phosphonic acid molecules have to be removed from the surface-functionalized particles. Coating of the oxide particles thus requires a number of additional preparation steps (synthesis of the phosphonic acid, the coating itself and washing of the coated particles), increasing the complexity and cost of the process. For this reason, it would be desirable to omit the surfactant treatment.
In this article, we investigate the morphology and dielectric properties of BaTiO3/poly(vinylidenfluoride-co-hexafluoro-propylene) (BTO/P(VDF-HFP)) films prepared by spin-coating. In contrast to earlier reports, the BTO nanoparticles were synthesized by a sol–gel technique, leading to a high density of surface hydroxyl groups.4,9,11,19,24,26 The effect of coating of these oxide particles with pentafluorobenzyl phosphonic acid (PFBPA) and the application of the dispersant BYK-W-9010 was investigated. We found that the untreated barium titanate leads to better film homogeneities and a large enhancement of the relative permittivity by a factor 2 compared to surface modified particles and even by a factor 10 compared to pure P(VDF-HFP). A very large relative permittivity εr of 57 was obtained at 1 kHz for a 50 vol% BaTiO3/P(VDF-HFP) composite with a low loss tangent in the order of 0.1. The possibility to avoid the coating of oxide particles strongly eases the preparation technique, making the obtained films more attractive for technical applications.
Experimental procedure
Material preparation
BaTiO3 (BTO) nanoparticles were synthesized by a sol–gel method starting from barium chloride dihydrate (60 mmol, BaCl2·2H2O, ≥99%, Sigma-Aldrich) and freshly distilled titanium(IV) chloride (60 mmol, TiCl4, 99.9% Acros Organics) in equimolar quantities as described in ref. 27. TiCl4 was hydrolysed in 30 mL ethanol followed by the dropwise addition of an aqueous solution of BaCl2·2H2O dissolved in 90 mL deionized and argon saturated water. Precipitation was achieved by fast addition of an excess potassium hydroxide solution (Sigma-Aldrich; 42 g in 100 mL water) and finally 100 mL water. The mixture was refluxed for 6 hours under argon atmosphere. The crystalline particles were washed three times with water, ethanol and acetone, respectively, and then dried in air.The phosphonic acid was synthesized by the Arbuzov reaction of 2,3,4,5,6-pentafluorobenzyl bromide (>99%, Sigma-Aldrich) and triethyl phosphite (>95%, Fluka).28 The obtained phosphonic ester was distilled in vacuum (150 °C, 3 mbar) to give the crude material as colourless oil. An excess of bromotrimethylsilane (97%, Sigma-Aldrich) was added for conversion to the corresponding silyl ester. The phosphonic acid was obtained by hydrolysis of the silyl ester with diluted hydrochloric acid (0.01 M). The surface functionalization was performed according to Kim et al.19 Typically 12 g oxide powder was dispersed in 300 mL ethanol (96%, analytical grade purity) for 6 hours by ultrasonication to prevent particle agglomeration. Afterwards, 6 mmol phosphonic acid dissolved in 50 mL ethanol was added and the mixture was again ultrasonicated for 1 hour at room temperature. The nanoparticles were separated by centrifugation and washed 3 times with excess ethanol under ultrasonication followed by centrifugation. Selected samples were additionally washed 3 times with water, afterwards with ethanol and finally with acetone to remove unreacted and physisorbed phosphonic acid as well as a water-dissoluble barium phosphonate byproduct described below. After washing, the particles were dried for 4 hours under vacuum at 80 °C. Poly(vinylidenfluoride-co-hexafluoropropylene) P(VDF-HFP) (average Mn ∼ 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich, containing 5% HFP) was used as purchased.
000, Sigma-Aldrich, containing 5% HFP) was used as purchased.
Preparation of composite films
Composite suspensions were prepared by ball-milling of P(VDF-HFP) dissolved in N,N-dimethylformamide (DMF, analytical grade purity) and the BTO nanoparticles for typically 4 h with 300 rpm in a planetary ball mill (Retsch PM 400). For selected batches, BYK-W 9010 (a phosphoric acid polyester copolymer with acid groups, BYK-Chemie) was added as additional dispersants prior to the ball-milling process. Composite films were fabricated by spin-coating on aluminum-coated substrates with 1000 rpm for 1 min. The aluminum electrode was prepared by thermal evaporation of 150 nm aluminum on a 24 mm × 24 mm glass substrate. The films were dried at room temperature in air and afterwards annealed at 50 °C for 20 minutes. Parallel-plate capacitors were obtained by deposing an array of 2 mm × 3 mm top aluminum electrodes (350 nm thickness) on the composite films through a mask using thermal evaporation (Vakuumtechnik Dresden model B-30 HVT) with 1 nm s−1 deposition rate.Measurements
Fourier transform infrared spectra with 2 cm−1 resolution were collected on a Bruker TENSOR spectrometer with attenuated total reflectance on a diamond crystal. Three-point BET gas physisorption measurements were done on a Quantachrome Corporation Nova 1000 using nitrogen as adsorbate in a range of p/p0 0.1–0.3 to calculate specific surface areas and averaged particle sizes. The powder density was determined by a Porotec Pycnomatic Helium-pycnometer at room temperature. Particle agglomeration was investigated with a Brookhaven Instruments Corporation Particle Size Analyzer 90 Plus. The data was averaged over three measurements. Thermal analysis including thermogravimetry (TG) and diffential thermal analysis (DTA) were performed in a Netzsch STA 449C system (heating rate 10 K min−1, Al2O3 crucibles, flowing air with 20 mL min−1). For transmission electron microscopy a Philips CM200 STEM was used. The film morphologies were investigated using an environmental scanning electron microscope (Philips ESEM XL 30 FEG). The elemental composition was analysed by coupled energy dispersive X-ray spectroscopy (EDX). Cross-sectional SEM images were prepared by fracturing liquid-nitrogen cooled films. The film thickness was additionally measured by means of a contact profilometer (Dektak 150 stylus surface profiler). Frequency-dependent capacitance and loss tangent values were obtained with an Agilent 4284A LCR meter in the range 120 Hz to 1 MHz at room temperature, where the voltage was kept constant at 1 V. All presented dielectric properties are averaged over four parallel-plate capacitors.Results and discussion
Characterization of as-prepared and surfactant-coated BTO particles
The properties of barium titanate are strongly influenced by particle size and applied synthesis route.29 In this article, we focus on a sol–gel synthesized powder consisting of spherical particles in the range of 20–50 nm as measured by TEM (Fig. 1). | ||
| Fig. 1 TEM bright-field image of BaTiO3 particles synthesized by the sol–gel procedure. | ||
The sol–gel soft-chemistry synthesis leads to a high concentration of surface hydroxyl groups which are good linkers for the surfactants. The particle size determined by TEM confirms the value for the crystallite size of 32 ± 4 nm calculated by the Scherrer equation using the (111) peak of the XRD measurements (see Fig. S1, ESI†). As described previously, the sol–gel synthesis of BaTiO3 leads to a highly crystalline product already at low reaction temperatures (i.e. without any calcination step) with a ratio of approximately 35% tetragonal and 65% pseudo-cubic phase as determined by Rietveld refinement.27 The formation of the pseudo-cubic phase is well known for very small BaTiO3 particles.30 Using a helium pycnometer, a density of 5.10 g cm−3 was obtained. This corresponds to roughly 85% of the crystallographic density indicating the dense nanoparticular structure and high crystallinity of the obtained powder. Furthermore, the density of our BaTiO3 samples is very similar to the one of commercially available nanopowders (e.g. Sigma-Aldrich Barium titanate(IV) nanopowder, <100 nm particle size (BET): 5.56 g cm−3).12,14
A fraction of the BTO powder was treated with 2,3,4,5,6-pentafluorobenzyl phosphonic acid (PFBPA), which is known to form stable bonds with the oxide surface, thus forming a surface layer with a good compatibility to the hydrophobic fluorinated polymer host. After surface treatment, stable BTO dispersions without sedimentation over several weeks in unpolar organic solvents were achieved. On the other hand, SEM investigations revealed the presence of an additional fraction of micrometer sized needles after the PFBPA treatment as shown in Fig. 2.
 | ||
| Fig. 2 SEM image of phosphonic acid surface treated powder on a Ba-phosphonate needle. | ||
EDX analysis of these needles revealed a high content of barium and also fluorine, which apparently stems from the phosphonic acid. It can therefore be concluded that the needles consist of barium phosphonate. In fact, it was possible to synthesize a Ba-phosphonate from Ba(OH)2·8H2O and PFBPA. This byproduct was found to be water soluble. Therefore, some samples were additionally washed with water after the surface treatment reaction (denoted as C-BTO@PFBPA in the following). Fig. 3 shows the thermogravimetric analysis (TG) of PFBPA-treated powders before and after the additional washing step.
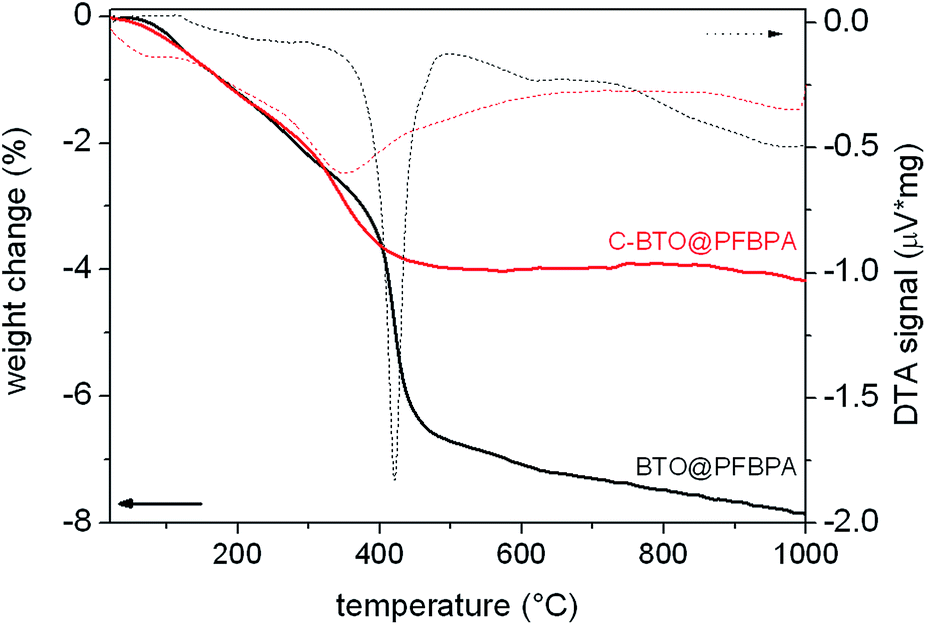 | ||
| Fig. 3 TG/DTA analysis of surface-coated powder before (BTO@PFBPA) and after (C-BTO@PFBPA) the water cleaning step. | ||
The thermogravimetry of BTO@PFBPA showed a significant weight loss between 350–500 °C accompanied by an exothermic signal in the differential thermal analysis (DTA). Mass spectrometry (not shown) revealed that this reaction step is correlated with the release of water, fluorine and carbon dioxide and can therefore be assigned to the oxidative degradation of the phosphonic acid chain, while phosphor remains in oxidic form in the sample.27 The weight loss of the washed sample C-BTO@PFBPA is much smaller and more diffuse (onset temperature 250 °C). For both samples the observed weight loss can be correlated with the BET specific surface area to calculate the degree of phosphonic acid surface coverage ΘPA, assuming a specific area of 0.24 nm2 per molecule phosphonic acid.31,32 In the corresponding calculations the weight change during the exothermic reaction was interpreted as the complete combustion of the organic chain of the phosphonic acid. In this case, the average number of bonded molecules per nm2 (PA) is given by
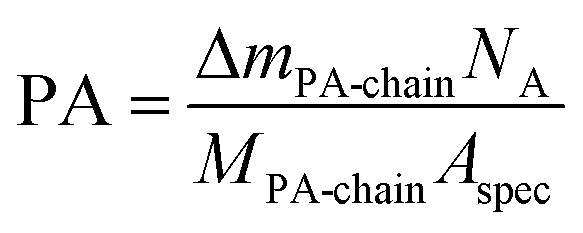 | (1) |
| Sample | A spec (BET) (m2 g−1) | Δm (%) | ΔTwt (°C) | PA (nm−2) | Θ PA (%) |
|---|---|---|---|---|---|
| BTO | 25.86 | ||||
| BTO@PFBPA | 20.45 | 4.51 | 350–500 | 7.33 | 176 |
| C-BTO@PFBPA | 23.06 | 2.81 | 250–500 | 4.05 | 97 |
The coverage higher than 100% for the BTO@PFBPA sample is striking. If the weight change is assigned to the loss of the entire phosphonic acid molecule as done e.g. in ref. 11, a surface coverage below 100% is obtained. However, mass spectrometry of the evolved gas gave no hints for phosphorous-containing volatile reaction products. For this reason, we assume that phosphor remains in the sample in oxidic form after decomposition and the weight change was assigned to the decomposition of the organic chain. One possible reason for a surface coverage larger than 100% is the formation of phosphonic acid multilayers.19,31,32 As an alternative explanation, we propose that the large value is due to the formation of the Ba-phosphonate shown in Fig. 2. This Ba-phosphonate may also form in commercial and/or thermally treated barium titanate powders by a leaching of barium ions, which is clearly more severe for the highly reactive sol–gel BTO. This interpretation is supported by the fact that after a thorough water cleaning step, the C-BTO@PFBPA sample showed a coverage of 97%, which corresponds to a monomolecular surface layer. Since the barium phosphonate is water soluble, it is reasonable to assume that the rinsing removes it completely.
FT-IR spectroscopy provides additional information on the surface modification products. PFBPA is known to form stable bonds to the BTO particle surface.11,19,22 The sol–gel soft-chemistry method applied in this work leads to nanoparticles with a high degree of surface hydroxyl groups, which are good linkers for the phosphonic acid.33Fig. 4 shows the FT-IR spectra of the surface-modified particles before and after the cleaning step in comparison with the pure BTO particles and Ba-phosphonate obtained from the reaction of Ba(OH)2·8H2O with the phosphonic acid.
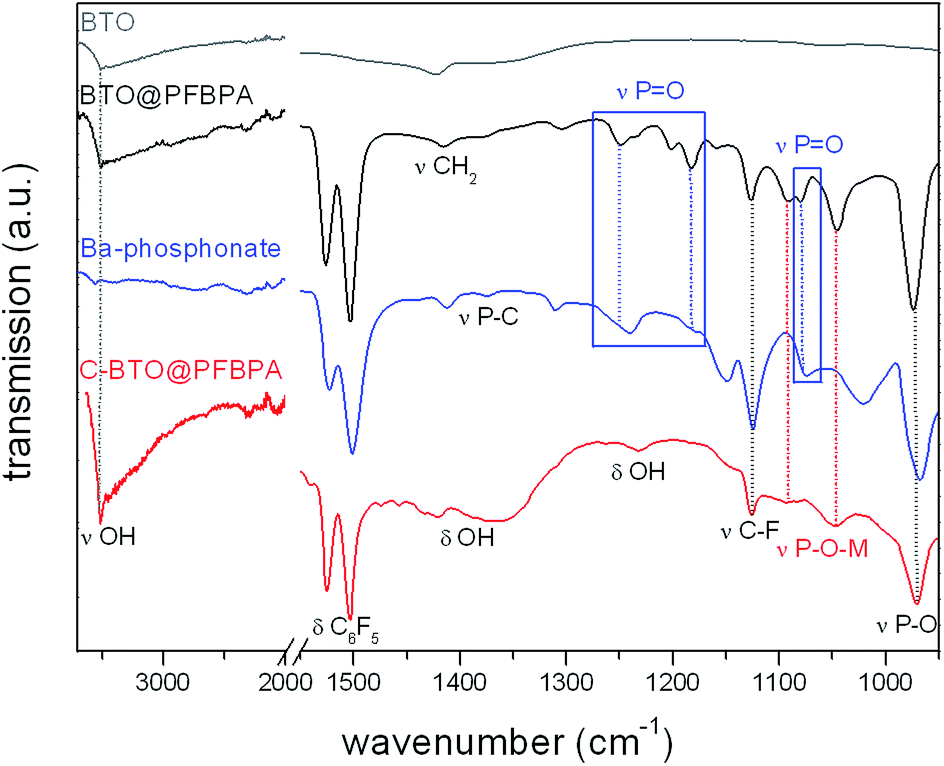 | ||
| Fig. 4 FT-IR spectra of pure BTO (grey), surface-modified BTO@PFBPA (black), isolated Ba-phosphonate byproduct (blue) and the purified C-BTO@PFBPA (red). | ||
All BTO samples exhibit the characteristic feature of remaining surface hydroxyl groups (3512 cm−1) which apparently cannot totally be saturated by the phosphonic acid. The broad P–OH stretching bands (around 2750 cm−1 and 2300 cm−1) are only found for pure PFBPA, but not for BTO@PFBPA, verifying that these groups form bonds to the oxide surface in at least a bidentate coupling mode.20,21,34 The comparison with the FT-IR spectra of PFBPA is shown in the ESI (Fig. S2†). After a typical surface functionalization, aromatic vibration modes (1526 cm−1 and 1502 cm−1) and the C–F stretching mode (1126 cm−1) were detected indicating the presence of PFBPA. In the finger-print region between 1300 cm−1 and 900 cm−1, major changes can be observed. Especially the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes (1258 cm−1 and 1072 cm−1) and the P–O stretching modes (1020 cm−1 and 968 cm−1) of the pure PFBPA (marked by dashed blue and black lines) collapses to two main signals of M–O–P vibration (M = metal; marked by dashed red lines at 1094 cm−1 and 1047 cm−1) in case of a successful surface bonding.11,19,22 After the surface modification, several additional signals remain around 1300–1200 cm−1 and 1080 cm−1 (marked by blue boxes), which can be attributed to the barium phosphonate. This byproduct can be eliminated by a washing step after the surface treatment. The resulting particles (C-BTO@PFBPA; bottom layer) show only signals, which can be attributed to the surface-bonded phosphonic acid, but also reveal the presence of a high content of adsorbed water and surface hydroxyl groups (around 1350 cm−1). It can be assumed that the large amount of hydroxyl groups causes an increase of dielectric loss and leakage current due to residual-free ionic groups.25 The high content of adsorbed water also prevents the polymer wetting of these particles, despite of the phosphonic acid coverage. As a consequence, the dielectric measurements of composites out of this powder showed a strong increase of the leakage current.
O stretching modes (1258 cm−1 and 1072 cm−1) and the P–O stretching modes (1020 cm−1 and 968 cm−1) of the pure PFBPA (marked by dashed blue and black lines) collapses to two main signals of M–O–P vibration (M = metal; marked by dashed red lines at 1094 cm−1 and 1047 cm−1) in case of a successful surface bonding.11,19,22 After the surface modification, several additional signals remain around 1300–1200 cm−1 and 1080 cm−1 (marked by blue boxes), which can be attributed to the barium phosphonate. This byproduct can be eliminated by a washing step after the surface treatment. The resulting particles (C-BTO@PFBPA; bottom layer) show only signals, which can be attributed to the surface-bonded phosphonic acid, but also reveal the presence of a high content of adsorbed water and surface hydroxyl groups (around 1350 cm−1). It can be assumed that the large amount of hydroxyl groups causes an increase of dielectric loss and leakage current due to residual-free ionic groups.25 The high content of adsorbed water also prevents the polymer wetting of these particles, despite of the phosphonic acid coverage. As a consequence, the dielectric measurements of composites out of this powder showed a strong increase of the leakage current.
For composite systems, the wetting interactions between oxide particles and the polymer host decide about the film homogeneity and the dielectric performance. The successful binding of the phosphonic acid is evidenced by hydrophobic properties of the coated BTO@PFBPA particles noticeable by the formation sedimentation-stable dispersions in different organic solvents. On the other hand, pure sol–gel BTO also formed stable particle dispersions in pure DMF and in the polymer solution without sedimentation over several weeks. This can be explained taking into account that the aprotic yet polar F atoms in the polymer chain enable strong interactions with the surface hydroxyl groups of the BTO nanoparticles. For a better insight into the dispersibility, the agglomeration behavior of pure and surface treated particles was investigated by Dynamic Light Scattering (DLS). We studied both the solvent-dispersed particles and the resulting composite suspensions, i.e. oxide particles plus polymer dispersed/dissolved in DMF. The results are depicted in Fig. 5.
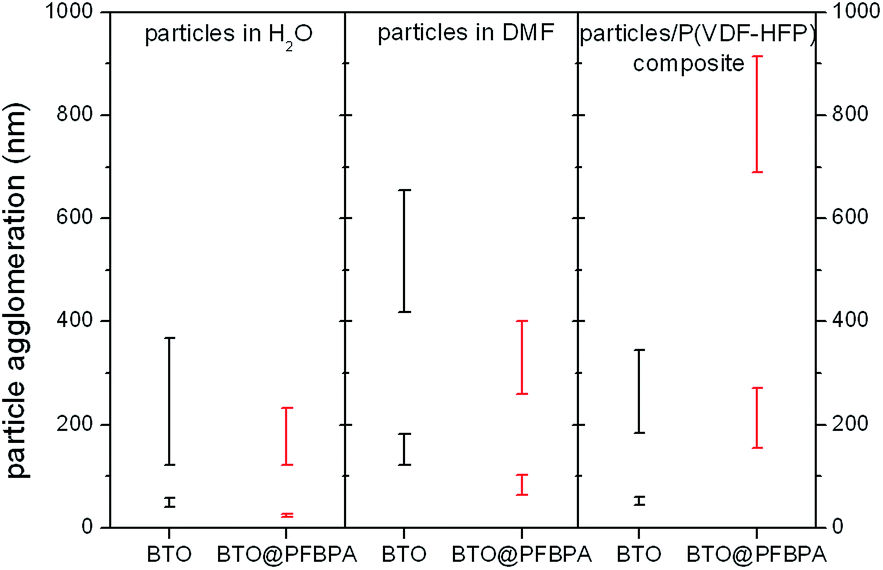 | ||
| Fig. 5 Particle agglomeration determined by DSL of pure sol–gel synthesized BTO and phosphonic acid surface modified BTO@PFBPA dispersed in water and DMF as well as in the resulting polymer composite suspension. Bars represent the variation of particle aggregate sizes. | ||
Typically, BTO nanoparticles tend to form large agglomerates in organic solvents or in composites because of their high surface energy and lipophobic properties resulting in severely inhomogeneous electric fields at the interfaces that can conduct charges and in turn reduce the dielectric breakdown strength.11,24,35 Therefore, numerous investigations focused on a suitable surface functionalization to suppress particle agglomeration and improve the polymer compatibility. Dynamic Light Scattering on such particles generally reveals a main fraction of almost isolated particles with a narrow size distribution and additionally a small fraction of much larger agglomerates. A similar particle size distribution was also observed in this work as shown in Fig. 5. In water the main fraction of BTO@PFBPA shows a slightly smaller particles size than pure BTO, what is surprising considering the hydrophobic properties of the surfactant. We assume that this effect is due to the long-term ultrasonication during the surface functionalization. On the other hand, an increasing particle agglomeration was found with progressing time before the DLS measurement. Therefore, the seemingly different sizes may simple be a time effect.
For both BTO and BTO@PFBPA, the size distribution of the agglomerates in DMF is slightly increased compared to water. On the other hand, the aggregation of BTO@PFBPA in DMF is obviously decreased compared to the pure BTO due to the hydrophobic surfactant layer.
The size distribution changes significantly when the polymer is added. It is to be expected that the fluorinated aromatic ring of the phosphonic acid creates a good chemical compatibility to the highly fluorinated chains of the chosen polymer.19,21 Nevertheless, we found that the aggregate sizes of our sol–gel BTO@PFBPA particles in the P(VDF-HFP)/DMF solution become larger than in pure DMF. As shown in the ESI† these aggregates are also slightly larger than the ones of phosphonic acid modified commercial BaTiO3 (Fig. S3†). Surprisingly, the pure (highly hydroxylated) sol–gel BTO particles form very small aggregates (<400 nm). This finding can be explained by the formation of hydrogen bonds between the surface OH-groups of the BTO nano particles and the F atoms of P(VDF-HFP), creating a good compatibility between BTO and polymer matrix. Such strong interactions have already been observed by Zhou et al.26 These authors activated the surface of BaTiO3 particles by H2O2 to introduce additional hydroxyl groups. This led to polymer composites with lower loss tangent and higher dielectric strength compared to the untreated material. It should be noted that the advantage of our approach is the formation of spherical nanoparticles with a high degree of surface hydroxyl groups in a simple one-step synthesis, i.e. without any surfactant functionalization. As expected, this good wetting leads to fewer voids in the composite, and in turn to a more homogeneous film morphology with better dielectric properties as described below.26
Morphology and dielectric properties of composite films
Thin films were prepared by spin-coating of BTO/polymer suspensions. Cross-section SEM was used to characterize the film morphologies. The particle distribution and overall homogeneity of the composite material are the key parameters for enhanced dielectric properties. We observed that C-BTO@PFBPA/P(VDF-HFP) composites show highly defective structures (voids, cracks, agglomerations) when water-rinsed powder was used. In turn, these composites exhibited a strong increase of the leakage current, which made it impossible to evaluate their dielectric properties. For this reason, these films are not further discussed in the following. In contrast, it was possible to prepare uniform composite thin films of BTO@PFBPA (i.e. containing the Ba-phosphonate byproduct). The resulting composite materials show isolated and uniformly distributed particles (Fig. 6). | ||
| Fig. 6 TEM bright-field image of a thin section of a BTO@PFBPA/P(VDF-HFP) composite film. | ||
Surprisingly, the large Ba-phosphonate needles found in the powder were not observed in the final composite films, most likely because the Ba-phosphonate is dissolved by DMF. Therefore, these needles do not interfere with the composite structure. More interestingly, it was found that homogeneous composite thin films can also be prepared out of the pure BTO powders, i.e. without phosphonic acid coating. Fig. 7 compares SEM images of both composite systems (phosphonic acid coated and pure BTO). A maximum oxide filling of 50 vol% was chosen as upper limit, which is near the percolation threshold evaluated by numerical calculations.11,24,36 For the high-content composites, the oxide particles are very densely packed, preventing a clear insight into the film morphology by SEM. Therefore, composites of 50 vol% and low-content composites of 10 vol% are presented for both BTO powders in Fig. 7.
 | ||
| Fig. 7 Cross-section SEM images of spin-coated phosphonic acid modified BTO@PFBPA/P(VDF-HFP) (left) and pure BTO/P(VDF-HFP) composites films (right) with different BaTiO3 volume fractions. | ||
For the BTO@PFBPA/P(VDF-HFP) composites regions with well-separated particles in the composite films were found but also regions with enriched oxide contents and polymer accumulations were identified, indicating an insufficient homogenization of the dispersions in spite of the surface coating by the phosphonic acid. Especially, the high content composite tended to form undesired blowholes. Against expectation, we observed that composites out of pure sol–gel BTO form layers with a clearly improved homogeneity. For pure BTO/P(VDF-HFP) composites the particles are well dispersed (as clearly visible in the image of the low oxide content composite) thus preventing percolation. In the high oxide content composite the particles are more densely packed similar to the situation in BTO@PFBPA/P(VDF-HFP), but without the unfavorable formation of flaws such as cracks or pores. This finding can be explained assuming that the small particle size and the highly hydroxylated BTO particle surface form a strong interaction with the F atoms of P(VDF-HFP), creating a good compatibility between BTO and polymer matrix.
Previous investigations revealed that an improved oxide-polymer wetting can also be achieved by additional dispersants such as BYK-W 9010 (BYK).37,38 BYK-W 9010 is known as a commercially available dispersion additive consisting of a phosphoric acid polyester copolymer with acid groups, It is frequently used to enhance the dispersability of oxide particles and was therefore tested in this study. Similar to other surfactants, it is supposed to link the particle surface with the polymer matrix. Therefore, we additionally investigated the effect of BYK with respect to the composite homogeneity and dielectric properties. Fig. 8 compares SEM images of both composite systems (phosphonic acid coated and pure BTO) with and without addition of BYK.
 | ||
| Fig. 8 Cross-section SEM images of composite films from phosphonic acid modified BTO@PFBPA/P(VDF-HFP) (left) and pure BTO/P(VDF-HFP) (right) with and without addition of the BYK dispersant. The BTO content was 30 vol% in all cases. | ||
As earlier reported, the use of BYK leads to a homogeneous particle distribution in the phosphonic acid modified BTO@PFBPA/P(VDF-HFP) system, resulting in an increase of the permittivity.27 As shown in the left part of Fig. 8, BYK effectively suppressed the formation of small defects. With this dispersant the particles are more uniformly distributed and surrounded by the polymer matrix, indicating that the phosphonic acid alone does not provide a sufficiently good connection between the oxide surface and the hydrophobic polymer. Surprisingly, the SEM imaged in the right part of Fig. 8 show that such morphological improvements are not achieved for the pure BTO/P(VDF-HFP) composites. In contrast even without BYK the BTO/P(VDF-HFP) films show a uniform and defect-free morphology. Both samples, i.e. with and without BYK, exhibited a similar particle distribution.
The SEM investigations are corroborated by the dielectric measurements discussed below, where a better homogeneity was found to lead to an increased permittivity (Fig. 9). In the dielectric measurements, the voltage was kept constant at 1 V and the obtained values were averaged over four parallel-plate capacitors. The results of the composite films are compared to a pure P(VDF-HFP) film, which was prepared in same way as the composite films.
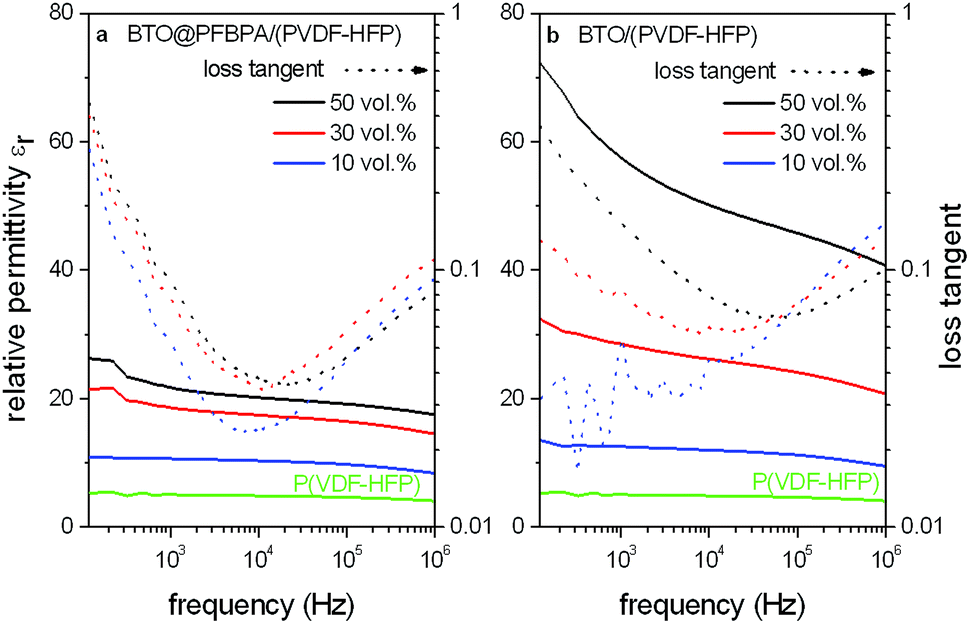 | ||
| Fig. 9 Relative permittivities and loss tangents of spin-coated phosphonic acid modified BTO@PFBPA/P(VDF-HFP) (a) and pure BTO/P(VDF-HFP) (b) composite films with different BaTiO3 volume fractions compared to the pure polymer. | ||
The observed relative permittivity of approximately εr ∼ 5 for P(VDF-HFP) is less than half of the specified value (εr ∼ 11.4) for the ideal crystalline polymer. This decrease can be explained by a reduced crystallinity due to the spin coating process without mechanically stretching.39,40 For the composite films of both the pure BTO and the phosphonic acid-coated oxide the relative permittivities εr strongly increase with rising oxide content. For the BTO/P(VDF-HFP) composite system out of untreated powder this increase is significantly larger. Comparing the permittivities of the composites with 50 vol% BTO to the pure P(VDF-HFP) polymer, an enhancement up to a factor of four for the phosphonic acid modified BTO@PFBPA/P(VDF-HFP) system was found.
On the other hand, for the pure BTO/P(VDF-HFP) system an even larger enhancement by a factor larger than 10 was achieved at 1 MHz with a further increase for lower frequencies. The loss tangents of phosphonic acid modified BTO@PFBPA/P(VDF-HFP) composites are almost independent of the oxide content, which is probably the result of the oxide surface treatment. Pure BTO/P(VDF-HFP) composites show a slightly increasing dielectric loss tangent with rising oxide filling at low frequencies. However, the values are not significantly higher than for the surface-coated oxide particles. Therefore, the use of pure BTO particles that stem from the sol–gel approach strongly enhances the dielectric permittivity while the loss tangent remains low.
Moreover, a reproducible effect of BYK on the permittivity of the pure BTO/P(VDF-HFP) composites was not observed (Fig. 10). Instead, it was found that the relative permittivity of BTO/P(VDF-HFP) composite films increases with BYK only for low oxide contents while it decreases for higher fillings. In contrast, the use of BYK in the phosphonic acid modified BTO@PFBPA/P(VDF-HFP) system leads to an increased homogeneity and permittivity.
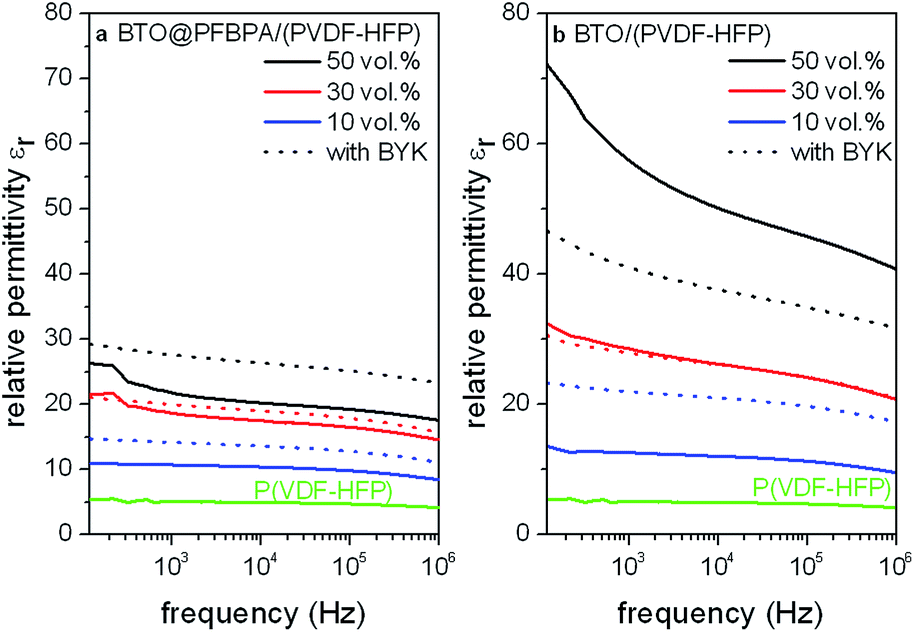 | ||
| Fig. 10 Relative permittivities of composite films from phosphonic acid modified BTO@PFBPA/P(VDF-HFP) (a) and pure BTO/P(VDF-HFP) (b) containing different BaTiO3 volume fractions with and without addition of the dispersant BYK-W 9010. | ||
These findings are supported by measurements of the film thicknesses and surface roughnesses. BTO@PFBPA/P(VDF-HFP) samples without additional dispersant show an increasing film thickness with higher oxide contents, typically ranging from 5.4(2) μm for the 10 vol% composites to 10.7(4) μm for 50 vol%. This effect is accompanied by an increasing surface roughness from approximately 0.1 μm to 0.4 μm. In contrast, the corresponding samples with BYK-W 9010 as additive exhibit uniform film thicknesses around 4.5(5) μm and surface roughnesses of 0.10(5) μm. This effect is not surprising because BYK reduces the viscosity of highly filled formulations by increasing the polymer wetting of particles. Additionally, we assume that the formation of the Ba-phosphonate during the functionalization of sol–gel BTO disturbs the surfactant–polymer interactions so that the phosphonic acid alone is not sufficient for linking the oxide surface and the polymer. In this case, BYK can effectively link both materials and increases the wettability, leading to uniform composite film thicknesses for various oxide contents.
It is noteworthy that the permittivities of pure BTO/P(VDF-HFP) samples were generally larger than those of the surface modified BTO@PFBPA/(PVDF-HFP) samples with or without BYK. Regarding the film thicknesses, the BTO/P(VDF-HFP) samples without additional dispersant exhibited very similar values around 4.2(4) μm and surface roughnesses of approximately 0.2 μm for all oxide contents, while samples with BYK-W 9010 as additive show decreasing film thicknesses with increasing oxide contents, typically ranging from approximately 4.6(6) μm for the 10 vol% composites to 2.9(3) μm for 50 vol%. The decreasing film thicknesses confirm an improved particle wetting by addition of the BYK dispersant. On the other hand, the decreasing film thicknesses worsen the relative permittivity. Especially the residual free acid groups of the dispersant cause an increase of dielectric loss and leakage current, which results in a negative effect for the relative permittivity of composites of pure and also functionalized BTO.25 These findings underline the positive influence of pure sol–gel BTO without surfactant-coating or additional dispersants on the composite homogeneity. The loss tangents of both BTO systems (phosphonic acid coated and pure BTO) with addition of BYK are almost identical compared to corresponding samples without BYK.
In this context it has to be noted that the dispersibility of BTO particles strongly depends on the preparation technique. Typical commercially available BTO particles do not possess a high degree of surface hydroxyl groups. Therefore, the surface modification of such BTO particles with phosphonic acids leads to a better dispersibility in the polymer solution. The corresponding composote systems with PVDF (or its copolymers) show relative permittivities in the range of 35–40, while composites without functionalization achieve slightly smaller relative permittivity up to 33.9,11,19 Previously, higher permittivities close to our values could only be realized in thick composite films or pellets from solution casting or hot-pressing.23,26,41–45 For our BTO nanoparticles from the precipitation method, coating with phosphonic acids was found to be not mandatory for a good dispersibility because the high amount of hydroxyl groups already generates a good wettability resulting in uniform particle distributions and enhanced dielectric properties.
In addition to the increased permittivity and low loss tangent of pure BTO/P(VDF-HFP) composites, we also found an enhancement of the dielectric strength by almost factor 2 compared to BTO@PFBPA/P(VDF-HFP) composites. While BTO@PFBPA/P(VDF-HFP) films exhibited dielectric strengths of approximately 90 V μm−1 (within a range of ±10 V μm−1), the BTO/P(VDF-HFP) composites afforded a significant improvement to approximately 170 V μm−1 (within a range of ±20 V μm−1). For neither composite films of the pure BTO nor for the phosphonic acid-coated oxide any systematic influence of the oxide content or additional dispersant on the dielectric strength could be identified.
Remarkably, our investigations show that when BTO nanoparticles prepared by a sol–gel route are applied as inorganic component, the best dielectric properties are achieved by keeping the composite system as simple as possible, i.e. without surfactant-coating of the oxide or application of additional dispersants. The permittivity of roughly 60 at 1 kHz is among the highest values ever reported for BTO-based nanocomposite dielectrics.4,9–11,19,26 The use of sol–gel BTO with a high degree of surface hydroxyl groups is therefore a clear advantage making the fabrication process much easier, faster and cheaper.
Conclusions
High-quality composite films were obtained by spin-coating of BaTiO3 embedded in a P(VDF-HFP) matrix. BaTiO3 synthesized by a sol–gel route appears as well-crystalline spherical nanoparticles with a high degree of surface hydroxyl groups, which are good linkers for surfactant molecules such as PFBPA, but also for the fluorinated polymer used as matrix material. Phosphonic acids are often used as detergents to form stable oxide surface bonds and to create a good connection to the fluorinated organic polymer host. However, we found that the surface treatment of the sol–gel synthesized BTO with PFBPA leads to the formation of a barium phosphonate. The surface-treated BTO@PFBPA powder exhibit an apparent surfactant coverage higher than 100% due to this barium phosphate byproduct, while the washed C-BTO@PFBPA powder led to a value of 97%, which corresponds to a complete monomolecular surface layer. Unfortunately, composites made from the washed powder showed severe morphological defects and a strong increase of the leakage current, precluding their application as dielectric material. In contrast, homogeneous composite film can be prepared using either unwashed BTO@PFBPA or pure BTO particles. Composite films of phosphonic acid modified BTO show some morphological defects. This formation of defects can be suppressed by the addition of BYK-W 9010 as dispersant. This indicates that the phosphonic acid itself is not sufficient for linking the oxide surface and the hydrophobic polymer.In contrast to previous investigations, in which mainly commercially available BTO nanoparticles were used, it was found that composites of sol–gel BTO form very homogeneous layers with well-separated particles even without an application of phosphonic acid or the additional dispersant BYK-W 9010. This result can be attributed to strong hydrogen bonds between the surface hydroxyl groups and the fluorine atoms of the polymer. These BTO/P(VDF-HFP) composite films show a doubling of the relative permittivity εr compared to films from PFBPA-functionalized particles and more than one order of magnitude higher values compared to the pure polymer. An extraordinarily high relative permittivity εr of 57 was obtained for a 50 vol% BaTiO3 composite at 1 kHz with a further increase for lower frequencies. In addition, relatively low loss tangents in the order of 0.1 and an enhanced dielectric strength of approximately 170 V μm−1 were found for these films.
According to our results a tremendous simplification of the fabrication process of composite films is possible. Highly homogenous composite films from sol–gel synthesized BTO can be obtained without any surface modifications or dispersants. This simple processability using a solution technique leads to films with superior dielectric properties. Therefore, these composites are promising candidates for film capacitors, e.g. in embedded applications or as multilayers for electrical energy storage.
Acknowledgements
The work was supported by the Federal Ministry for Education and Research (BMBF) within the ForMaT/SuperKon grant. For density measurements, we thank Jenny Bienias from the group of Prof. Dr Thomas Hahn. Additional thanks are due to the German Federal Environmental Foundation (DBU) for financial support of a PhD scholarship.Notes and references
- P. Barber, S. Balasubramanian, Y. Anguchamy, S. Gong, A. Wibowo, H. Gao, H. J. Ploehn and H.-C. zur Loye, Materials, 2009, 2, 1697–1733 CrossRef CAS PubMed.
- T. J. Lewis, J. Phys. D: Appl. Phys., 2005, 38, 202–212 CrossRef CAS.
- P. Murugaraj, D. Mainwaring and N. Mora-Huertas, J. Appl. Phys., 2005, 98, 054304 CrossRef PubMed.
- H.-I. Hsiang, K.-Y. Lin, F.-S. Yen and C.-Y. Hwang, J. Mater. Sci., 2001, 36, 3809–3815 CrossRef CAS.
- H. Xu, Y. Bai, V. Bharti and Z.-Y. Cheng, J. Appl. Polym. Sci., 2001, 82, 70–75 CrossRef CAS.
- H. T. Vo and F. G. Shi, Microelectron. J., 2002, 33, 409–415 CrossRef CAS.
- N. Joseph, S. K. Singh, R. K. Sirugudu, V. R. K. Murthy, S. Ananthakumar and M. T. Sebastian, Mater. Res. Bull., 2013, 48, 1681–1687 CrossRef CAS PubMed.
- E. Tuncer, B. Nettelblad and S. M. Gubañski, J. Appl. Phys., 2002, 92, 4612–4624 CrossRef CAS PubMed.
- I. Vrejoiu, J. D. Pedarnig, M. Dinescu, S. Bauer-Gogonea and D. Bäuerle, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 407–409 CrossRef CAS.
- Y. Kobayashi, T. Tanase, T. Tabata, T. Miwa and M. Konno, J. Eur. Ceram. Soc., 2008, 28, 117–122 CrossRef CAS PubMed.
- P. Kim, N. M. Doss, J. P. Tillotson, P. J. Hotchkiss, M. J. Pan, S. R. Marder, J. Li, J. P. Calame and J. W. Perry, ACS Nano, 2009, 3, 2581–2592 CrossRef CAS PubMed.
- T. Hanemann, H. Gesswein and B. Schumacher, Microsyst. Technol., 2011, 17, 195–201 CrossRef CAS PubMed.
- T. Hanemann and B. Schumacher, Microsyst. Technol., 2012, 18, 745–751 CrossRef CAS PubMed.
- B. Schumacher, H. Geßwein, J. Haußelt and T. Hanemann, Microelectron. Eng., 2010, 87, 1978–1983 CrossRef CAS PubMed.
- G. Chen and A. E. Davies, IEEE Trans. Dielectr. Electr. Insul., 2000, 7, 401–407 CrossRef CAS.
- Y. Shen, Y. Lin, M. Li and C.-W. Nan, Adv. Mater., 2007, 19, 1418–1422 CrossRef CAS.
- J. K. Nelson and J. C. Fothergill, Nanotechnology, 2004, 15, 586 CrossRef CAS.
- D. Ma, T. A. Hugener, R. W. Siegel, A. Christerson, E. Mårtensson, C. Önneby and L. S. Schadler, Nanotechnology, 2005, 16, 724 CrossRef CAS.
- P. Kim, S. C. Jones, P. J. Hotchkiss, J. N. Haddock, B. Kippelen, S. R. Marder and J. W. Perry, Adv. Mater., 2007, 19, 1001–1005 CrossRef CAS.
- P. H. Mutin, G. Guerrero and A. Vioux, J. Mater. Chem., 2005, 15, 3761–3768 RSC.
- T. Schulmeyer, S. A. Paniagua, P. A. Veneman, S. C. Jones, P. J. Hotchkiss, A. Mudalige, J. E. Pemberton, S. R. Marder and N. R. Armstrong, J. Mater. Chem., 2007, 17, 4563–4570 RSC.
- S. A. Paniagua, P. J. Hotchkiss, S. C. Jones, S. R. Marder, A. Mudalige, F. S. Marrikar, J. E. Pemberton and N. R. Armstrong, J. Phys. Chem. C, 2008, 112, 7809–7817 CAS.
- J. Li, J. Claude, L. E. Norena-Franco, S. I. Seok and Q. Wang, Chem. Mater., 2008, 20, 6304–6306 CrossRef CAS.
- S. Siddabattuni, T. P. Schuman and F. Dogan, Mater. Sci. Eng., B, 2011, 176, 1422–1429 CrossRef CAS PubMed.
- S.-D. Cho and K.-W. Paik, in Electronic Components and Technology Conference, 2001. Proceedings., 51st, 2001, pp. 1418–1422 Search PubMed.
- T. Zhou, J.-W. Zha, R.-Y. Cui, B.-H. Fan, J.-K. Yuan and Z.-M. Dang, ACS Appl. Mater. Interfaces, 2011, 3, 2184–2188 CAS.
- C. Ehrhardt, C. Fettkenhauer, J. Glenneberg, W. Münchgesang, C. Pientschke, T. Großmann, M. Zenkner, G. Wagner, H. S. Leipner, A. Buchsteiner, M. Diestelhorst, S. Lemm, H. Beige and S. G. Ebbinghaus, Mater. Sci. Eng., B, 2013, 178, 881–888 CrossRef CAS PubMed.
- A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415–430 CrossRef CAS.
- N. Nuraje and K. Su, Nanoscale, 2013, 5, 8752–8780 RSC.
- S. Yoon, S. Baik, M. G. Kim, N. Shin and I. Kim, J. Am. Ceram. Soc., 2007, 90, 311–314 CrossRef CAS PubMed.
- W. Gao, L. Dickinson, C. Grozinger, F. G. Morin and L. Reven, Langmuir, 1996, 12, 6429–6435 CrossRef CAS.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367–4373 CrossRef CAS.
- S.-J. Chang, W.-S. Liao, C.-J. Ciou, J.-T. Lee and C.-C. Li, J. Colloid Interface Sci., 2009, 329, 300–305 CrossRef CAS PubMed.
- I. Gouzman, M. Dubey, M. D. Carolus, J. Schwartz and S. L. Bernasek, Surf. Sci., 2006, 600, 773–781 CrossRef CAS PubMed.
- R. A. Vaia and J. F. Maguire, Chem. Mater., 2007, 19, 2736–2751 CrossRef CAS.
- J. P. Calame, J. Appl. Phys., 2006, 99, 084101 CrossRef PubMed.
- A. G. Pedroso, D. S. Rosa and T. D. Z. Atvars, J. Appl. Polym. Sci., 2004, 92, 1834–1839 CrossRef CAS.
- Z. Amjad, Phosphorus Res. Bull., 2006, 20, 159–164 CrossRef CAS.
- M. Wegener, W. Kunstler, K. Richter and R. Gerhard-Multhaupt, J. Appl. Phys., 2002, 92, 7442–7447 CrossRef CAS PubMed.
- A. C. Jayasuriya and J. I. Scheinbeim, Appl. Surf. Sci., 2001, 175–176, 386–390 CrossRef CAS.
- Z.-M. Dang, H.-Y. Wang, Y.-H. Zhang and J.-Q. Qi, Macromol. Rapid Commun., 2005, 26, 1185–1189 CrossRef CAS.
- M.-F. Lin, V. K. Thakur, E. J. Tan and P. S. Lee, RSC Adv., 2011, 1, 576–578 RSC.
- H.-J. Ye, W.-Z. Shao and L. Zhen, Colloids Surf., A, 2013, 427, 19–25 CrossRef CAS PubMed.
- K. Yu, H. Wang, Y. Zhou, Y. Bai and Y. Niu, J. Appl. Phys., 2013, 113, 034105 CrossRef PubMed.
- B.-H. Fan, J.-W. Zha, D.-R. Wang, J. Zhao, Z.-F. Zhang and Z.-M. Dang, Compos. Sci. Technol., 2013, 80, 66–72 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD pattern of BTO obtained by the sol–gel synthesis. FT-IR spectra of pure BTO and PFBPA as well as the surface-modified BTO@PFBPA and isolated Ba-phosphonate byproduct and the purified C-BTO@PFBPA. Detailed investigations of the particle agglomeration determined by DSL of pure sol–gel synthesized BTO and phosphonic acid surface modified BTO@PFBPA and modified commercially available nanopowders BTOcom@PFBPA (Sigma-Aldrich Barium titanate(IV) nanopowder, <100 nm particle size (BET)) dispersed in water, DMF and in the resulting polymer composite suspension. See DOI: 10.1039/c4ra03715d |
| This journal is © The Royal Society of Chemistry 2014 |