
Fabrication and characterization of flexible silk fibroin films reinforced with graphene oxide for biomedical applications
Abstract
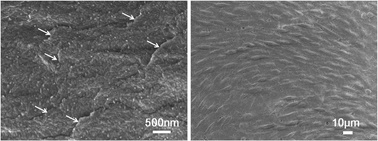
Nanocomposite films based on silk fibroin (SF) and graphene oxide (GO) were fabricated by simply casting the two components in aqueous media. The structure and properties of the films were systematically characterized by scanning electron microscope (SEM), X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), differential scanning calorimetry (DSC), thermal gravimetric analysis (TGA) and tensile tests. The investigations demonstrated that the presence of GO in the SF matrix increased the silk II content in the composite films by the intermolecular forces between GO and SF molecular chains. These newly formed interactions also contributed to the improvement of the film's mechanical properties and thermal stability. Further, the biodegradation behavior and biocompatibility were investigated to evaluate the biological performance of the SF/GO films. Compared with the SF film, the films prepared by incorporating GO showed a lower enzymatic degradation rate, reflecting their improved resistance to the applied enzyme solution. The cell viability and proliferation results in vitro indicated that the composite films, especially the SF/GO film with 0.5 wt% GO, could support cell adhesion and proliferation. This study has developed a novel, flexible and biocompatible SF/GO composite film by a green and feasible method, which should encourage the development of a series of SF/GO-based materials for biomedical applications.
 Please wait while we load your content...
Please wait while we load your content...

