
Simulation of conformational properties of end-grafted diblock copolymers
Abstract
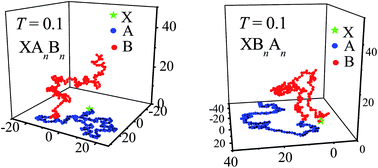
Conformational properties of end-grafted flexible diblock copolymers were studied by using Monte Carlo simulation. The copolymers, XAnBn and XBnAn, are grafted with the end X to a flat surface which attracts monomers A but repulses monomers B. Results show that the blocks A in XAnBn and XBnAn are adsorbed upon the surface at low temperature, and the adsorption of blocks A takes place roughly at the same temperature. However, the conformational size and instantaneous shape of the blocks A and B in XAnBn and XBnAn are different. The possible reasons were discussed and they were interpreted by different properties and grafting locations of the two blocks.
 Please wait while we load your content...
Please wait while we load your content...

