Probing structure-functionality relationships of catalytic bimetallic Pt–Ru nanoparticles associated with improved sulfur resistance†
Jessica N. G. Stanley,
Paul Benndorf,
Falk Heinroth,
Anthony F. Masters and
Thomas Maschmeyer*
Laboratory of Advanced Catalysis for Sustainability, School of Chemistry F11, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: thomas.maschmeyer@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 4504
First published on 16th June 2014
Abstract
Supported bimetallic Pt–Ru alloyed nanoparticulate catalysts, which show improved sulfur tolerance for a model aqueous phase reforming reaction, were investigated using X-ray absorption spectroscopy (XANES and EXAFS) at the Pt LIII and the Ru K-edges, before and after exposure to thiophene, a sulfur-containing model poison. Preliminary EXAFS investigations confirmed the alloyed character of the bimetallic catalysts, and further experiments allowed us to clearly determine more detailed changes to the Pt–Ru bonding environments as induced by sulfur poisoning, i.e. partial particle dealloying. However, after treating the poisoned catalysts with pure H2 at 300 °C, the Pt–Ru alloy appeared to regenerate. These results, based on the atomic environments of the absorbing species, are consistent with our previous catalytic and bulk powder X-ray diffractive investigations, and support our proposed sulfur and hydrogen spillover in situ self regeneration mechanism.
Introduction
Biomass is one of the most promising renewable sources for liquid fuels and chemicals.1–5 Due to the differences in oxygen and moisture contents, thermal stabilities, and functionality between biomass and petroleum feedstocks, new chemical approaches are required for biomass processing, and catalysts are involved in many of the processes.6,7 A promising technology for biomass conversion is aqueous phase reforming (APR).8 APR uses supported metal catalysts at relatively low temperatures and pressures for the conversion of biomass-derived feedstocks to hydrogen,9–15 gaseous alkanes,13,16,17 and liquid alkanes,18–23 and the products can be tailored by altering the catalyst and H2/CO concentrations in the reactor. However, it is expected that the performance of the catalysts will be impaired by the sulfur present in real biomass feedstocks (e.g., already for wood it is approx. 56 ppm, significant protein content worsens this situation further, the sulfur substantially deriving from the amino acids cysteine and methionine and the uptake of soil nutrients),24 as sulfur is well-known to poison noble metal catalysts.25,26 Thus, there is a great need to develop sulfur-resistant catalysts for biomass conversions.We envisage the operation of a combined sulfur and hydrogen spillover mechanism on supported bimetallic catalysts to overcome these issues. Bimetallic catalysts are used in industrial applications as they often show improved activity, selectivity, and sulfur resistance compared to their monometallic counterparts.11,27–30 This improved performance could result from metal–metal bonding or changes to the number of available active sites on the surface of the catalyst leading to electronic perturbations, or edge-sites, kinks, or defects resulting in structural changes.30 The concept of a sulfur ‘spillover’ was proposed by Azad and Duran for Pd–Re catalysts that have applications in the steam reforming of sulfur-laden logistic fuels (diesel, gasoline, jet fuel).31,32 They suggested that the catalyst could remain active for longer as palladium is able to transfer sulfur onto the more readily sulfidable rhenium. Although we acknowledge that ‘sulfur spillover’ is only loosely connected to the extensively studied conventional hydrogen spillover mechanism,33,34 given the literature precedent, we adopt the same terminology for sulfur as well.
To facilitate these two spillover mechanisms on the same catalyst, we chose platinum for its hydrogenation ability and ruthenium, as it has a greater affinity for sulfur than does platinum.35 Thus, any sulfur species that binds to platinum could spillover to ruthenium. Similarly, hydrogen could spillover from platinum to the sulfur species bound to ruthenium, and cleave a sulfur-containing derivative, effectively regenerating the catalyst. Note that these spillover mechanisms are most likely to occur if the platinum and ruthenium are separated only by atomic dimensions, e.g., if they exist in the same lattice, perhaps as an alloy or a solid solution. A schematic of the proposed in situ self regeneration pathway is shown in Fig. 1.
 | ||
| Fig. 1 Schematic of the proposed in situ self regeneration mechanism, showing thiophene as sulfur poisoning model compound. | ||
Indeed, we have designed bimetallic Pt–Ru catalysts, supported on mesoporous SiAlTUD-1, that exhibit improved sulfur resistance and promising potential for the APR of model compounds, using the hydrogenation of cyclohexene to cyclohexane as a screening reaction.36 These bimetallic catalysts achieve turnover frequencies higher than their monometallic counterparts in both the absence and presence of sulfur-containing species (as thiophene). What is more remarkable is that while the monometallic catalysts are completely poisoned, the bimetallic catalysts remain active even in the presence of sulfur concentrations that are ten times that expected in woody biomass feedstocks – a concentration that is equivalent to a sulfur![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal molar ratio of 17:1 for the bimetallic catalyst with the highest metal loading, Pt1Ru1.5 (of course, not all metal atoms are exposed, so the ratio of sulfur to active metal species is much higher). Thus, these catalytic results are completely consistent with the operation of the proposed sulfur and hydrogen spillover mechanism on the bimetallic catalysts.
:metal molar ratio of 17:1 for the bimetallic catalyst with the highest metal loading, Pt1Ru1.5 (of course, not all metal atoms are exposed, so the ratio of sulfur to active metal species is much higher). Thus, these catalytic results are completely consistent with the operation of the proposed sulfur and hydrogen spillover mechanism on the bimetallic catalysts.
X-ray diffraction (XRD) characterisation of the bimetallic catalysts suggests that platinum and ruthenium atoms are incorporated in the same lattice, which is required for the operation of the regeneration mechanism. Further XRD studies of the used Pt1Ru1.5 catalyst suggest changes to the Pt unit cell size after poisoning, indicated by the shifting of the Pt reflections and changes to the Pt lattice constant. These changes are consistent with the catalyst losing some of its defined alloy character, partially separating into two amorphous and maybe somewhat elementally distinct domains. Thus, the XRD analyses suggest that the observed restructuring of the metal nanoparticles results from increasing amounts of sulfur-containing species being co-ordinated to the ruthenium atoms during poisoning, leading to a change in alloy structure. Regeneration of the catalyst in pure hydrogen at 300 °C shows this to be a reversible process.
Although the catalytic results and the XRD analysis are consistent with the proposed regeneration pathway, extended X-ray absorption fine structure (EXAFS) was needed to probe the alloyed character of the bimetallic catalysts at the atomic level (in terms of nearest neighbour environments), and to provide further insights into the regeneration mechanism. Moreover, EXAFS was required to afford structural information on the bimetallic catalysts for which the peak shifts in the XRD were slight or not apparent, and to examine the possible contribution(s) of amorphous phases. The fingerprint of the X-ray absorption near edge structure (XANES) spectra is also useful in qualitatively determining the metallic character of the catalysts.
Herein, we report the XANES and EXAFS results of experiments confirming the alloyed character of Pt1.5Ru1, Pt1Ru1.5, and Pt1Ru3 bimetallic catalysts, and the EXAFS results of experiments probing the sulfur poisoning and regeneration mechanism of Pt1.5Ru1 and Pt1Ru3 catalysts.
Results and discussion
The synthesis, general characterisation, and catalytic activity of Pt, Ru, Pt–Ru, Pt1Ru3, Pt1Ru1.5, and Pt1.5Ru1 mono- and bimetallic catalysts have been described previously.36XANES
The XANES spectra of the unused catalysts at the Pt LIII-edge and Ru K-edge are shown in Fig. 2. The spectra of the reference materials (Pt foil, Ru foil, PtO2, and RuO2) are included for comparison with those of the catalysts. At the Pt LIII-edge (Fig. 2a), the absorption positions of the catalysts were similar to the Pt foil, suggesting a highly metallic character. On the other hand, at the Ru K-edge (Fig. 2b), some oxidation was suggested for the Pt1Ru1.5 catalyst, as shown by the flat top between the two characteristic absorptions of metallic ruthenium. The oxidation of ruthenium is not surprising, as ruthenium is oxophillic,37 and no precautions to exclude air were taken when handling the catalysts. However, this contrasts with the Ru XANES spectra of the other catalysts, which are similar to that of the Ru foil, suggesting that they have predominantly metallic character. The higher metal loading of the Pt1Ru1.5 catalyst compared to the other two bimetallic catalysts could account for why oxidation was only readily observed for this catalyst.36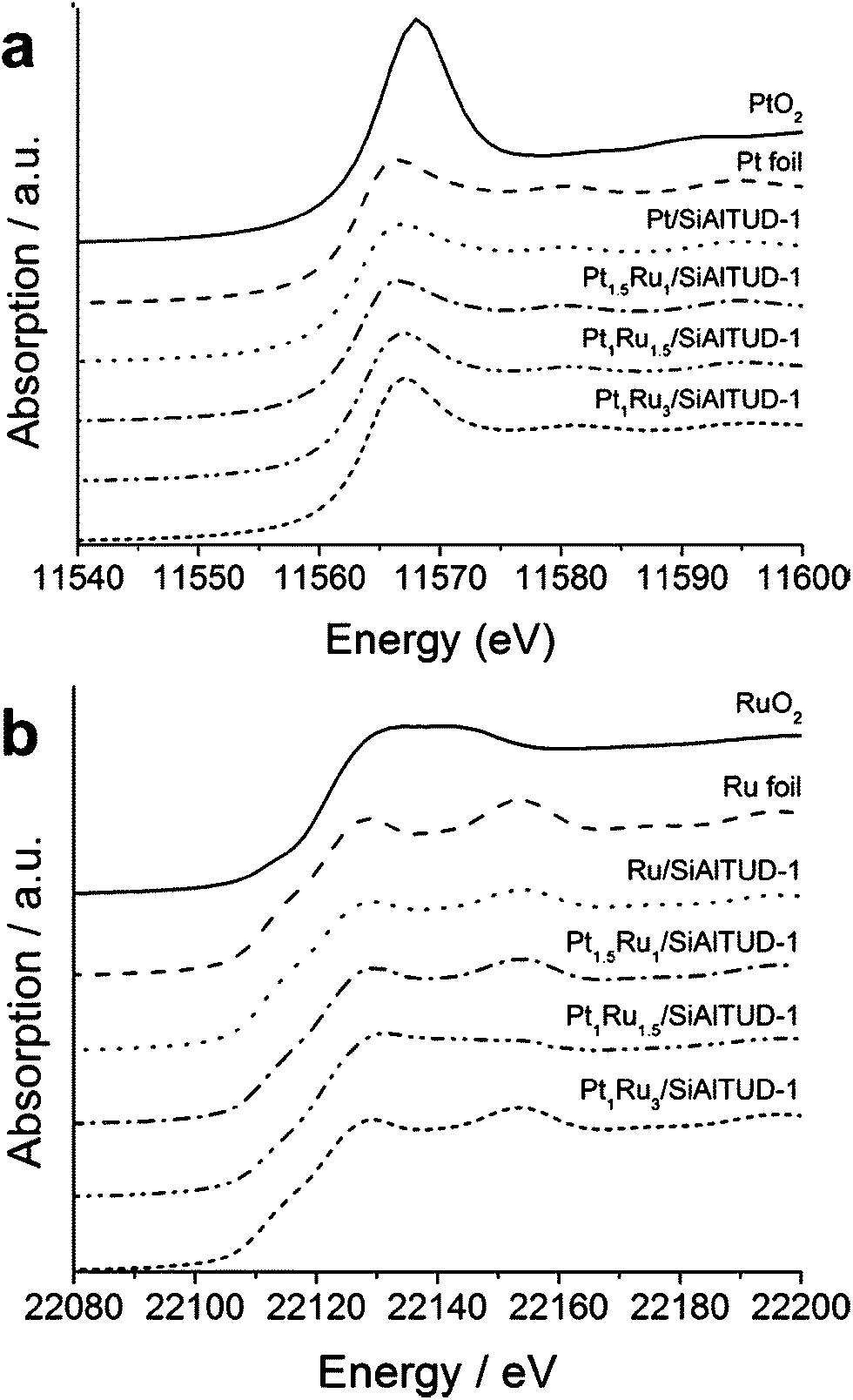 | ||
| Fig. 2 XANES spectra at the (a) Pt LIII-edge and (b) Ru K-edge for the unused catalysts compared with the reference samples. | ||
EXAFS
 | ||
| Fig. 3 (a) Raw χ(k) × k3 EXAFS data and (b) Fourier transformed EXAFS data from Pt foil measured at the Pt LIII-edge. Observed (solid line), calculated (short dashed line), residual (dotted line), and window (long dashed line). | ||
 | ||
| Fig. 4 (a) Raw χ(k) × k3 EXAFS data and (b) Fourier transformed EXAFS data from Ru foil measured at the Ru K-edge. Observed (solid line), calculated (short dashed line), residual (dotted line), and window (long dashed line). | ||
| Pt LIII-edge | |||||||
|---|---|---|---|---|---|---|---|
| Pt foil | Monometallic Pt catalyst | ||||||
| R (Å) | N | σ2 (Å2) | R (Å) | N | σ2 (Å2) | ||
| Pt–Pt | 2.76 | 12f | 0.001 | Pt–Pt | 2.76 | 8.9 | 0.003 |
| Pt–Pt | 3.91 | 6f | 0.002 | Pt–Pt | 3.91 | 4.4 | 0.004 |
| Pt–Pt | 4.80 | 24f | 0.002 | Pt–Pt | 4.80 | 15.9 | 0.004 |
| Pt–Pt | 5.53 | 12f | 0.003 | Pt–Pt | 5.53 | 8.9 | 0.006 |
| R-factor = 5.81% | Chi2 = 0.57 | R-factor = 8.22% | Chi2 = 0.53 | ||||
| Ru K-edge | |||||||
|---|---|---|---|---|---|---|---|
| Ru foil | Monometallic Ru catalyst | ||||||
| R (Å) | N | σ2 (Å2) | R (Å) | N | σ2 (Å2) | ||
| a Restrained where N5 < 12. | |||||||
| Ru–Ru | 2.67 | 12f | 0.002 | Ru–Ru | 2.67 | 8.3 | 0.002 |
| Ru–Ru | 3.79 | 6f | 0.002 | Ru–Ru | 3.79 | 4.4 | 0.002f |
| Ru–Ru | 4.25 | 2f | 0.001 | Ru–Ru | 4.26 | 0.5 | 0.0009 |
| Ru–Ru | 4.67 | 18f | 0.003 | Ru–Ru | 4.67 | 6.1 | 0.0004 |
| Ru–Ru | 5.08 | 12f | 0.001 | Ru–Ru | 5.08 | 12.5a | 0.003 |
| Ru–Ru | 5.33 | 16f | 0.003 | Ru–Ru | 5.33 | 6.6 | 0.0008 |
| R-factor = 11.0% | Chi2 = 2.15 | R-factor = 10.3% | Chi2 = 1.41 | ||||
Good fits were obtained for the Pt and Ru foils, as shown in Fig. 3 and 4, and as suggested by the low R-factors and chi2 values reported for the best-fit parameters in Table 1 and 2, at the Pt LIII-edge and Ru K-edge, respectively. The refined bond distances were all consistent with the corresponding crystal structures.38 It is worth noting that six shells were required to obtain a good fit for the Ru foil, as a model using five shells previously reported in the literature for the same range was inadequate in fitting the present data.37
Similarly, good fits were obtained for the Pt and Ru monometallic catalysts (best-fit parameters reported in Table 1 and 2, respectively). For the Ru catalyst, it was necessary to fix the Debye–Waller factor in the second shell to 0.002 σ2, which was the value obtained for the Debye–Waller factor in the first Ru shell, as a physically meaningful value could not be obtained when this parameter was free to vary. Fixing this parameter resulted in only a slight increase of the R-factor, from 8.69% to 10.3%, and the chi2 value decreased, from 2.10 to 1.41. Thus, as the R-factor did not significantly change, the chi2 value was low, and all the other parameters were sensible, the discrepancy of one Debye–Waller factor can be considered to be within the limitations of the model. The Pt LIII- and Ru K-edge models determined for these reference samples were subsequently used as the basis for the models of the three bimetallic catalysts.
The discrepancies between the small nanoparticle sizes calculated from the first shell co-ordination numbers obtained by EXAFS and the previous characterisation could arise as the values obtained from EXAFS represent an average of all the metal atoms present in the catalyst, i.e., crystalline and non-crystalline domains, whereas the XRD results include only crystalline domains. Furthermore, at these very small sizes the Scherrer equation is edging towards its limits of applicability and as only the relatively larger nanoparticles are detected by XRD this biases the calculation of the particle sizes. Similarly, the three dimensional pore structure of the SiAlTUD-1 support also makes it very difficult to distinguish the smaller nanoparticles by TEM inspection.
| Pt LIII-edge | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt1.5Ru1 | Pt1Ru1.5 | Pt1Ru3 | |||||||||
| R (Å) | N | σ2 (Å2) | R (Å) | N | σ2 (Å2) | R (Å) | N | σ2 (Å2) | |||
| Pt–Pt | 2.76 | 9.9 | 0.0008 | Pt–Pt | 2.76 | 5.9 | 0.001 | Pt–Pt | 2.75 | 3.1 | 0.001 |
| Pt–Pt | 3.91 | 4.9 | 0.001 | Pt–Pt | 3.91 | 2.5 | 0.001 | Pt–Pt | 3.90 | 1.7 | 0.003 |
| Pt–Pt | 4.80 | 16.6 | 0.001 | Pt–Pt | 4.79 | 7.8 | 0.002 | Pt–Pt | 4.78 | 3.0 | 0.0009 |
| Pt–Pt | 5.53 | 9.9 | 0.003 | Pt–Pt | 5.53 | 5.9 | 0.004 | Pt–Pt | 5.53 | 0.4 | 0.001 |
| Pt–Ru | 2.72 | 1.2 | 0.009 | Pt–Ru | 2.70 | 0.3 | 0.001 | Pt–Ru | 2.70 | 2.2 | 0.002 |
| Pt–Ru | 3.79 | 0.3 | 0.0009 | ||||||||
| Pt–Ru | 4.70 | 3.9 | 0.007 | ||||||||
| Pt–Ru | 5.41 | 2.2 | 0.005 | ||||||||
| R-factor = 4.96% | Chi2 = 1.45 | R-factor = 5.78% | Chi2 = 0.50 | R-factor = 9.67% | Chi2 = 2.41 | ||||||
| Ru K-edge | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt1.5Ru1 | Pt1Ru1.5 | Pt1Ru3 | |||||||||
| R (Å) | N | σ2 (Å2) | R (Å) | N | σ2 (Å2) | R (Å) | N | σ2 (Å2) | |||
| Ru–Ru | 2.68 | 7.8 | 0.003 | Ru–Ru | 2.68 | 2.7 | 0.002 | Ru–Ru | 2.67 | 8.5 | 0.003 |
| Ru–Ru | 3.79 | 2.4 | 0.0008 | Ru–Ru | 3.78 | 1.3 | 0.0003 | Ru–Ru | 3.79 | 2.8 | 0.0007 |
| Ru–Ru | 4.26 | 0.2 | 0.001 | Ru–Ru | 4.23 | 1.9 | 0.005 | Ru–Ru | 4.29 | 3.1 | 0.001 |
| Ru–Ru | 4.68 | 7.0 | 0.002 | Ru–Ru | 4.68 | 3.4 | 0.003 | Ru–Ru | 4.67 | 6.2 | 0.0009 |
| Ru–Ru | 5.05 | 4.1 | 0.0008 | Ru–Ru | 5.07 | 5.5 | 0.002 | Ru–Ru | 5.08 | 12.2 | 0.004 |
| Ru–Ru | 5.34 | 4.2 | 0.0006 | Ru–Ru | 5.34 | 2.6 | 0.01 | Ru–Ru | 5.34 | 9.4 | 0.002 |
| Ru–Pt | 2.72 | 2.3 | 0.005 | Ru–Pt | 2.70 | 3.4 | 0.006 | Ru–Pt | 2.70 | 0.2 | 0.001 |
| Ru–O | 2.00 | 0.7 | 0.001 | ||||||||
| R-factor = 5.84% | Chi2 = 0.64 | R-factor = 16.6% | Chi2 = 1.41 | R-factor = 6.17% | Chi2 = 1.74 | ||||||
The best fit for the Pt1.5Ru1 catalyst required the addition of one Pt–Ru shell at the Pt LIII-edge and one Ru–Pt shell at the Ru K-edge (Table 3 and 4, respectively). The first shell bond distance for the Pt–Ru and Ru–Pt shells were 2.72 Å at both edges, which is intermediate between the values for the bond distances of Pt–Pt (2.76 Å) and Ru–Ru (2.68 Å). The value for the heteroatom bond distance suggests the contracting of the Pt host lattice by the smaller Ru atoms, which provides further evidence for alloy formation.
The best fit for the Pt1Ru3 catalyst required four Pt–Ru shells at the Pt LIII-edge and one Ru–Pt shell at the Ru K-edge (Table 3 and 4, respectively). The first shell bond distance for the Pt–Ru and Ru–Pt shells in this case was 2.70 Å at both edges, suggesting the expansion of the Ru host lattice by the larger Pt atoms. At the Pt LIII-edge, the bond distances obtained for the second to fourth Pt–Ru shells also suggested an expansion of the Ru host lattice. The bond distances obtained for all Pt–Ru shells were consistent with those reported by Nashner et al. for simultaneously fitted Pt LIII-edge and Ru K-edge EXAFS data obtained for carbon-supported [PtRu5]/C nanoparticles.45
In the case of the Pt1Ru1.5 catalyst, the inclusion of an Ru–O shell (with a Ru–O bond distance of 2.0 Å) in addition to an Ru–Pt shell was required at the Ru K-edge (Table 4), consistent with the oxidation suggested in the XANES spectrum for this catalyst (Fig. 2b). At the Pt LIII-edge, one Pt–Ru shell was needed. The Pt–Ru and Ru–Pt bond distances were 2.70 Å, which is smaller than the Pt–Ru and Ru–Pt bond distances obtained for the Pt1.5Ru1 catalyst, and is consistent with the incorporation of more Ru atoms into the Pt host lattice of Pt1Ru1.5 compared with the Pt1.5Ru1 catalyst.
The EXAFS investigation into the sulfur poisoning and regeneration mechanism was continued for the Pt1.5Ru1 and Pt1Ru3 catalysts. As there are relatively fewer Ru atoms in Pt1.5Ru1 compared to Pt1Ru1.5, the shifting in lattice parameters observed by XRD is only slight, while in the case of the Pt1Ru3 catalyst, no shifting of peak positions in the XRD pattern is observed, likely because of the noisy data. For these two catalysts, EXAFS experiments were performed on fresh, poisoned, and regenerated catalyst samples. ‘Fresh’ refers to unused catalyst. ‘Poisoned’ refers to the catalyst that was used in the model hydrogenation reaction in the presence of 450 ppm of sulfur and then dried at room temperature in air. ‘Regenerated’ refers to the catalyst that was used in the presence of 450 ppm of sulfur, dried at room temperature in air, and heated for 2 h under hydrogen at 300 °C.
The raw and Fourier transformed EXAFS data for Pt1Ru3 at the Pt LIII-edge are shown in Fig. 5. Qualitative analysis of the Fourier transformed data suggested differences between the poisoned sample and the fresh and regenerated samples. The dominant peak at ∼2.5 Å in the Fourier transform results from contributions of both Pt and Ru scatterers. In the poisoned sample, the shoulder to the right of the peak disappeared, but was present in the regenerated sample. Thus, changes to the sample during poisoning appear to take place and can be followed with atomic resolution (in terms of nearest neighbour), and these changes are reversed (in terms of what can be gleaned via EXAFS analysis and catalytic performance data) after the regeneration of the catalyst.
 | ||
| Fig. 5 Raw χ(k) × k3 EXAFS data for the (a) fresh, (b) poisoned, and (c) regenerated sample and Fourier transformed EXAFS data for the (d) fresh, (e) poisoned, and (f) regenerated sample of the Pt1Ru3 catalyst measured at the Pt LIII-edge. Observed (solid line), calculated (short dashed line), residual (dotted line), and window (long dashed line). | ||
Quantitative analysis of the EXAFS data for both the Pt1Ru3 and Pt1.5Ru1 catalysts provided further insights into structural changes to the Pt and Ru bonding environments induced by sulfur poisoning and re-activation, and provided evidence at the atomic level that a dealloying/re-alloying mechanism was occurring. The poisoned and regenerated samples were fitted starting with the parameters obtained for the fresh Pt1Ru3 and Pt1.5Ru1 catalysts, reported in Table 3 and 4 (above). As the focus of the analysis was on changes to the co-ordination numbers between the different samples (i.e. fresh, poisoned, and regenerated samples) for each catalyst, the Debye–Waller factors and passive electron amplitude reduction factor (S02) were fixed to the values obtained for the fresh samples, as the co-ordination numbers were found to be highly correlated with these parameters. All other parameters were free to vary. Tables with the complete best-fit parameter sets for both catalysts can be found in the ESI.† Note that as EXAFS shows the average of all the metal atoms, contributions from atoms not involved in the alloying are not discriminated against. This means that the overall effect is less pronounced compared to the clear changes in the Pt unit cell that could be observed for Pt1Ru1.5 by XRD.36 Thus, while the overall effect is evident in EXAFS, it is diluted compared to the effect visible in XRD, since the XRD effectively filters out all amorphous particles.
The analyses of the results for the Pt1Ru3 and Pt1.5Ru1 catalysts are shown in Fig. 6 and 7, respectively. The graphs depict the relative percentage of Pt and Ru metal in the first shell for fresh, poisoned, and regenerated samples, at both the Pt LIII-edge (Fig. 6a and 7a) and Ru K-edge (Fig. 6b and 7b). The first shell co-ordination numbers only were used in the calculation as a single metal heteroatom shell was modelled in all but one case.
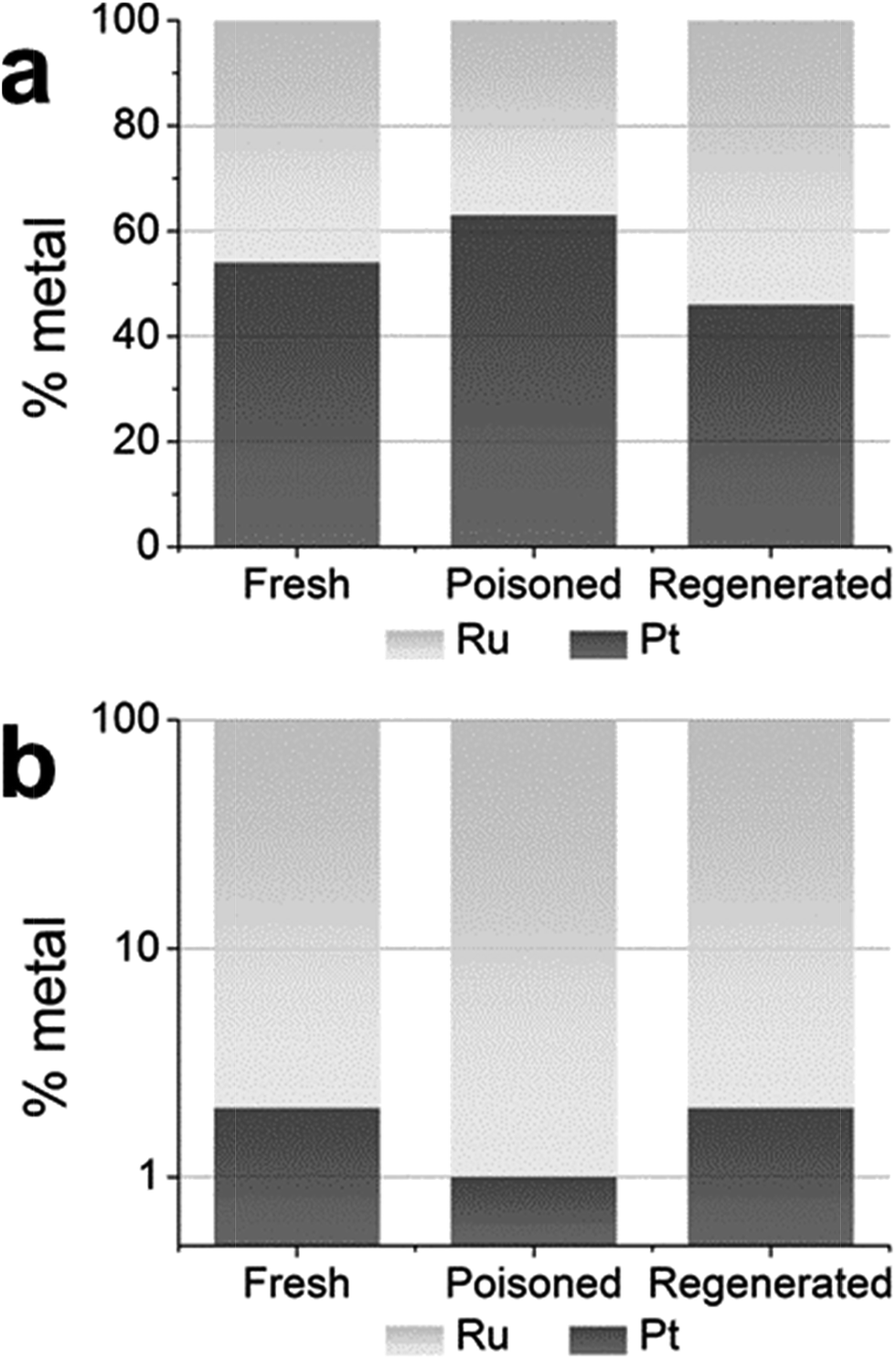 | ||
| Fig. 6 Relative percentages of Pt and Ru metal in the first shell of fresh, poisoned, and regenerated samples of the Pt1Ru3 catalyst, calculated for the (a) Pt LIII-edge and (b) Ru K-edge. | ||
 | ||
| Fig. 7 Relative percentages of Pt and Ru metal in the first shell of fresh, poisoned, and regenerated samples of the Pt1.5Ru1 catalyst, calculated for the (a) Pt LIII-edge and (b) Ru K-edge. | ||
Examining the graph for the Pt1Ru3 catalyst at the Pt LIII-edge (Fig. 6a), it is clear that the relative percentage of platinum increased after sulfur poisoning, while the relative percentage of ruthenium decreased. That is, after sulfur poisoning there were relatively more Pt–Pt bonding environments and relatively fewer Pt–Ru bonding environments. Conversely, at the Ru K-edge (Fig. 6b), after sulfur poisoning there were relatively more Ru–Ru bonding environments and relatively fewer Ru–Pt bonding environments. These results are consistent with the catalyst dealloying during poisoning, which could be due to the ruthenium being complexed to sulfur residues and the platinum separating out of the lattice. This situation was reversed when the catalyst was regenerated, which is consistent with the sulfur no longer being complexed to the ruthenium, and the platinum being reincorporated into the ruthenium host lattice.
Although the effect is only subtle (see below), similar trends were seen in the poisoned samples for the Pt1.5Ru1 catalyst (Fig. 7), suggesting that this catalyst was also partially dealloying during sulfur poisoning. At the Pt LIII-edge (Fig. 7a), there were slightly more Pt–Pt bonding environments compared to Pt–Ru environments after sulfur poisoning, while the opposite was true at the Ru K-edge (Fig. 7b). However, although it is clear at the Ru K-edge that the catalyst had re-alloyed after regeneration, this was not evident at the Pt LIII-edge. Indeed, the XRD data for the regenerated sample suggested the existence of an extra platinum phase. As platinum is a much stronger scatterer than ruthenium, this additional platinum phase could make it difficult to see the Pt–Ru bonding environments using EXAFS analysis, which appears to be the case with this sample.
While it was previously suggested that a correlation could exist between the presence of the alloy character after poisoning and the activity of the catalysts,36 a similar trend could not be determined in the present case. Nonetheless, these results suggested that the sulfur resistance could be improved if, during poisoning, a catalyst was more readily able to retain its alloy character, which could be achieved through the more efficient operation of the spillover mechanism. If there is more ruthenium available to act as a sulfur trap, sulfur-resistance could be improved, but if the ruthenium ratio is too high compared with platinum, the spillover mechanism could be hindered.
Experimental section
Catalyst preparation
The syntheses, general characterisation by ICP-AES, XRD, TEM, and nitrogen sorption measurements, and catalytic activity of the Pt, Ru, Pt1.5Ru1, Pt1Ru1.5, and Pt1Ru3 mono- and bimetallic catalysts have been described elsewhere.36 The XAS studies described below were measured using the same samples to facilitate the continued investigation into the sulfur poisoning mechanism. In addition, a Pt foil, Ru foil (both EXAFS Materials), PtO sample (Johnson Matthey), and RuO2 sample (Aldrich), were used as reference materials to inform and aid the modelling of the catalytic samples.X-ray absorption spectroscopy
The XAS experiments were performed at the wiggler XAS beamline at the Australian Synchrotron, Victoria, Australia. Spectra of the catalytic samples, and the PtO and RuO2 samples, were recorded at the Pt LIII-edge (11580 eV) and at the Ru K-edge (22135 eV), at ∼12 K in fluorescence mode with a 100-element solid-state Ge detector (Canberra). The scan range extended to k = 18 Å (EXAFS) with a step size of 0.01 Å at the pre-edge region, 0.00025 Å at the edge region, and 0.035 Å at the EXAFS region. Reference spectra of the Pt and Ru foils were measured in transmission mode at ∼12 K at the Pt LIII-edge and Ru K-edge, respectively.Samples measured at the Pt LIII-edge were pre-cooled in liquid nitrogen before being cooled and maintained at ∼12 K using a closed-cycle He cryostat (Cryo Industries), whereas samples measured at the Ru K-edge were cooled directly in the cryostat. The total data acquisition time per scan was approximately 45–60 min, and repeat scans (up to a total of three scans per sample) were acquired to improve the signal-to-noise ratios of the scans. Thus, the data reported represent averages of up to three individual acquisitions, with a total collection time of up to 3 h per data set.
The effect of sulfur poisoning and regeneration on the Pt–Ru bonding environments was investigated by measuring fresh, poisoned, and regenerated catalyst samples. ‘Fresh’ refer to catalysts that have not been used in any catalytic reaction. ‘Poisoned’ samples were obtained by performing an hydrogenation reaction (described previously) in the presence of thiophene (0.20 μL, in 1.0 mL ethanol, equivalent to a thiophene content of 450 ppm (v/v), or a sulfur content of 560 ppm (mol mol−1) in 160 μL cyclohexene.)36 The poisoned samples were subsequently isolated from the reaction mixture by centrifugation, and dried at room temperature in air. ‘Regenerated’ samples were poisoned and isolated as described above, before being heat-treated under H2 for 1 h at 300 °C (ramp rate 5 °C min−1).
Catalytic samples were ground using an agate mortar and pestle to ensure homogeneity, before being packed into a PMMA multi-slit sample holder (slit size 2 × 5 mm) sealed with Kapton tape. PtO and RuO2 were diluted with cellulose (1 wt%), similarly ground, and packed into the sample holder.
Calibration, averaging, and normalisation of XAS data were performed using the Average and Spline programs within the XFit software package.46 Fittings of EXAFS data were performed using the XFit software based on the FEFF6 code.47
Conclusions
We are able to confirm the alloyed character of three bimetallic catalysts that show improved sulfur tolerance in the presence of thiophene for a model APR reaction. Analyses of the first shell EXAFS best-fit parameters of the Pt1Ru3 and Pt1.5Ru1 catalysts clearly shows that the metal partially dealloyed during poisoning, which we suggest is due to the ruthenium being sequestered by the sulfur species. However, after regenerating the poisoned catalysts under hydrogen, the sulfur is removed and the alloy is reformed. Evidence that this re-alloying could occur is suggested by the EXAFS analyses of the regenerated catalysts. Thus, these results provide evidence that the in situ self regeneration mechanism, proposed earlier,36 occurs indeed, i.e. during poisoning the metal partially dealloys, but a sulfur spillover and a hydrogen spillover take place to regenerate the catalyst in situ, and the platinum and ruthenium are then re-alloyed, enabling the catalyst to remain active, and, significantly, sulfur-tolerant.Acknowledgements
We thank the Australian Research Council for support. Th. M. acknowledges the receipt of an ARC Future Fellowship (FT0990485) and DP grant (DP0987166), JNGS acknowledges the receipt of an Australian Postgraduate Award. This research was undertaken on the XAS beamline at the Australian Synchrotron, Victoria, Australia (AS131/XAS/5923 and AS132/XAS/6471). Preliminary research was undertaken at the Australian National Beamline Facility at the Photon Factory in Japan, operated by the Australian Synchrotron (AS113/ANBF/4185 and AS113/ANBF/4193). We acknowledge the Australian Research Council for financial support and the High Energy Accelerator Research Organisation (KEK) in Tsukuba, Japan, for operations support. We thank Dr Bernt Johannessen (AS) and Dr Jade Aitkin (ANBF) for their assistance with the XAS experiments, and Dr Aviva Levina for help with preliminary data analysis.Notes and references
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- D. L. Klass, Biomass for Renewable Energy, Fuels, and Chemicals, Academic Press, San Diego, 1998 Search PubMed.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777–793 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- L. D. Schmidt and P. J. Dauenhauer, Nature, 2007, 447, 914–915 CrossRef CAS PubMed.
- A. Demirbas, Prog. Energy Combust. Sci., 2007, 33, 1–18 CrossRef CAS PubMed.
- G. W. Huber and J. A. Dumesic, Catal. Today, 2006, 111, 119–132 CrossRef CAS PubMed.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222, 180–191 CrossRef CAS PubMed.
- J. W. Shabaker, R. R. Davda, G. W. Huber, R. D. Cortright and J. A. Dumesic, J. Catal., 2003, 215, 344–352 CrossRef CAS.
- G. W. Huber, J. W. Shabaker, S. T. Evans and J. A. Dumesic, Appl. Catal., B, 2006, 62, 226–235 CrossRef CAS PubMed.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2078 CrossRef CAS PubMed.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2003, 43, 13–26 CrossRef CAS.
- R. R. Davda and J. A. Dumesic, Chem. Commun., 2004, 36–37 RSC.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- Y.-C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 CAS.
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS PubMed.
- R. M. West, Z. Y. Liu, M. Peter, C. A. Gartner and J. A. Dumesic, J. Mol. Catal. A: Chem., 2008, 296, 18–27 CrossRef CAS PubMed.
- R. M. West, Z. Y. Liu, M. Peter and J. A. Dumesic, ChemSusChem, 2008, 1, 417–424 CrossRef CAS PubMed.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L.-t. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gaertner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59–70 CrossRef CAS PubMed.
- J. M. Robinson, S. R. Barrett, K. Nhoy, R. K. Pandey, J. Phillips, O. A. Ramirez and R. I. Rodriguez, Energy Fuels, 2009, 23, 2235–2241 CrossRef CAS.
- C. H. Bartholomew, P. K. Agrawal and J. R. Katzer, Adv. Catal., 1982, 31, 135–242 CAS.
- J. A. Rodriguez, Prog. Surf. Sci., 2006, 81, 141–189 CrossRef CAS PubMed.
- J. A. Rodriguez and D. W. Goodman, Surf. Sci. Rep., 1991, 14, 1–107 CrossRef CAS.
- G. A. Somorjai, Introduction to Surface Chemistry and Catalysis, Wiley, New York, 1994 Search PubMed.
- H. Yasuda and Y. Yoshimura, Catal. Lett., 1997, 46, 43–48 CrossRef CAS.
- J. A. Rodriguez and J. Hrbek, Acc. Chem. Res., 1999, 32, 719–728 CrossRef CAS.
- A.-M. Azad and M. J. Duran, Appl. Catal., A, 2007, 330, 77–88 CrossRef CAS PubMed.
- A.-M. Azad and D. Sundararajan, Adv. Mater. Sci. Eng., 2010, 325612, DOI:10.1155/2010/325683.
- W. C. Conner, Jr and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef.
- F. Roessner, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2008, vol. 3, pp. 1574–1585 Search PubMed.
- H. Wise, in Catalyst Deactivation Proceedings of the 5th International Symposium, ed. C. H. Bartholomew and J. B. Butt, Elsevier Science Publishers, Amsterdam, 1991, vol. 68, pp. 497–503 Search PubMed.
- J. N. G. Stanley, K. Worthington, F. Heinroth, A. F. Masters and T. Maschmeyer, Catal. Today, 2011, 178, 164–171 CrossRef CAS PubMed.
- J. Teddy, A. Falqui, A. Corrias, D. Carta, P. Lecante, I. Gerber and P. Serp, J. Catal., 2011, 278, 59–70 CrossRef CAS PubMed.
- A. W. Hull, Phys. Rev., 1921, 17, 571–588 CrossRef CAS.
- R. E. Benfield, J. Chem. Soc., Faraday Trans., 1992, 88, 1107–1110 RSC.
- Z. J. B. A. D. Van, D. C. Koningsberger, B. H. F. J. Van't and D. E. Sayers, J. Chem. Phys., 1985, 82, 5742–5754 CrossRef PubMed.
- P. Gallezot, A. I. Bienenstock and M. Boudart, Nouv. J. Chim., 1978, 2, 263–266 CAS.
- M. Vaarkamp, J. T. Miller, F. S. Modica and D. C. Koningsberger, J. Catal., 1996, 163, 294–305 CrossRef CAS.
- Y. Lei, J. Jelic, L. C. Nitsche, R. Meyer and J. Miller, Top. Catal., 2011, 54, 334–348 CrossRef CAS.
- J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, J. Catal., 2006, 240, 222–234 CrossRef CAS PubMed.
- M. S. Nashner, A. I. Frenkel, D. L. Adler, J. R. Shapley and R. G. Nuzzo, J. Am. Chem. Soc., 1997, 119, 7760–7771 CrossRef CAS.
- XFit for Windows, beta version, Australian Synchrotron Research Program, Sydney, 2004.
- J. J. Rehr, R. C. Albers and S. I. Zabinsky, Phys. Rev. Lett., 1992, 69, 3397–3400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Models; best-fit parameters of fresh, poisoned, and regenerated samples; and lists of restraints. See DOI: 10.1039/c4ra03474k |
| This journal is © The Royal Society of Chemistry 2014 |