Lithium deposition on graphite anode during long-term cycles and the effect on capacity loss
Lijie Yanga,
Xinqun Cheng*a,
Yunzhi Gaoa,
Yulin Maa,
Pengjian Zuoa,
Chunyu Dua,
Yingzhi Cuia,
Ting Guana,
Shuaifeng Loua,
Fuping Wanga,
Weidong Feib and
Geping Yin*a
aInstitute of Advanced Chemical Power Sources, School of Chemical Engineering and Technology, Harbin Institute of Technology, Harbin 150001, China. E-mail: chengxq@hit.edu.cn; yingeping@hit.edu.cn; Fax: +86-451-86413717; Tel: +86-451-86413707
bSchool of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150001, China
First published on 23rd May 2014
Abstract
Lithium deposition on the surface of a graphite anode during long-term cycles was evaluated using a LiCoO2/graphite battery. The batteries were charged/discharged at 1 C and 25 °C within the voltage range of 2.75–4.2 V for 600, 700, 800, 900 and 1000 cycles. Scanning electron microscopy (SEM) results indicated that both solid electrolyte interphase (SEI) film and lithium deposition appeared on the surface of the cycled graphite anode. Dendritic and granular lithium deposits grew on the anode non-uniformly. Metallic lithium existed in the deposition according to differential scanning calorimetry (DSC) results. Capacity declined distinctly from the 800th cycle, corresponding with the growth of lithium deposits. An SEI film was formed on the surface of the lithium deposits. Results of X-ray photoelectron spectroscopy (XPS) test indicated that the composition of SEI film on the surface of the lithium deposits was the same as that of the SEI film on the surface of cycled graphite. Capacity loss from the electrolyte consumed by the formation of the SEI film was 23.61%, while the loss from other battery components was 76.39%. Formation of lithium deposits consumed active lithium in the battery and led to capacity loss. According to test results of the three-electrode cell, the average anode potential at the end of constant-current charging for full battery became more negative with the cycling, and this phenomenon was related to the generation of lithium deposits.
Introduction
The known reasons for capacity loss upon charging/discharging cycles of LiCoO2/graphite batteries in the voltage range of 2.75–4.2 V are the continuous growth of a solid electrolyte interphase (SEI) film on the surfaces of the cathode1,2 and anode,3,4 the transformation of structures5–8 and so on.The cathode and anode may sometimes not be charged and discharged during the ideal voltage range even though the full battery was cycled normally at 2.75–4.2 V. S. S. Zhang et al.9 found that potential of the graphite anode could be lower than 0 V (vs. Li+/Li) during the constant-current charging of the full battery. Therefore, it was possible for lithium to be deposited on the graphite surface. The phenomenon may be more severe when the battery was charged at larger current or at lower temperature. The charging properties of the batteries could be improved by increasing the capacity ratio of the anode to cathode (A/C) appropriately. Mao-Sung Wu et al.10 reported that the potential of the mesocarbon microbead (MCMB) electrode decreased to −0.1 V (vs. Li+/Li) during charging when the A/C was 0.9. Gray deposition appeared on the surface of the fully charged MCMB, but it did not appear for the battery with an A/C of 1.05. However, most of these studies focused on the effect of A/C on lithium deposition during the early stage of the cycling. If the deposits were gradually generated on the surface of the carbon during long-term charge/discharge cycles, the process would also affect the capacity loss of the battery before leading to safety problems,11–13 which has not yet been studied clearly. In this experiment, the phenomenon of lithium deposition in a long-term cycled battery, its effect on capacity loss of the battery and the mechanism of deposition formation were evaluated.
Experimental
The Li-ion battery used in this experiment was a commercial cell with a nominal capacity of 1000 mA h. The cathode was prepared by coating the mixing slurry, which consisted of LiCoO2, acetylene black, BP2000, polyvinylidene fluoride (PVDF) and a certain amount of N-methyl-2-pyrrolidinone (NMP), on aluminum foil. The anode consisted of graphite, Super-P, styrene–butadience rubber (SBR) and carboxymethyl cellulose (CMC), and the electrolyte was 1 mol L−1 LiPF6 dissolved in ethylene carbonate–diethyl carbonate–ethyl methyl carbonate (EC–DEC–EMC, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 by volume). Vinylene carbonate (VC) and propylene sulfite (PS) as electrolyte additives were used in the commercial lithium ion battery. The batteries were charged and discharged at 0.2 C first to obtain the capacity and then cycled at 1 C for 600, 700, 800, 900 and 1000 times. Capacity verification of cycled batteries was conducted at 0.2 C. All the charging and discharging was performed at 25 °C within the range of 2.75–4.2 V.
:1:1 by volume). Vinylene carbonate (VC) and propylene sulfite (PS) as electrolyte additives were used in the commercial lithium ion battery. The batteries were charged and discharged at 0.2 C first to obtain the capacity and then cycled at 1 C for 600, 700, 800, 900 and 1000 times. Capacity verification of cycled batteries was conducted at 0.2 C. All the charging and discharging was performed at 25 °C within the range of 2.75–4.2 V.
The cycled batteries were dismantled in a glove box filled with argon gas. The obtained anodes were immersed in dimethyl carbonate (DMC) to remove the electrolyte LiPF6. The morphology and composition of anodes were examined by scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). In order to avoid exposure to air and moisture, the testing samples were sealed in argon-filled vessels and then transferred to SEM and XPS instruments. Atomic force microscopy (AFM) analysis was conducted on different areas of the graphite anode cycled 1000 times (Bruker Dimension Icon). The sample for differential scanning calorimeter (DSC, NETZSCH, STA 449 F3 Jupiter) analysis was obtained from the cycled graphite anode with the deposition and was sealed in a gold sample pan in the glove box. The heating temperature ranged from 30 °C to 230 °C and the heating rate was 10 °C min−1.
In order to examine the effect of cycled electrolyte on capacity loss, fresh electrolyte was added into the cycled batteries and the obtained battery was charged and discharged at 0.2 C to examine the capacity.
The cell after charge/discharge cycles was dismantled in the glove box and the obtained plate group including the cathode, anode and separator were put into fresh electrolyte, along with the metal lithium plate reference electrode (RE). The RE was close enough to the anode and was isolated from the anode by a separator layer. The sealed three-electrode cell is shown in Fig. 1. The electrode potential of the graphite anode (vs. Li+/Li) was monitored when the LiCoO2/graphite battery was charged and discharged at 1 C within the range of 2.75–4.2 V.
 | ||
| Fig. 1 Diagram of the three-electrode cell of the LiCoO2/graphite battery. | ||
Results and discussion
LiCoO2/graphite batteries were charged and discharged for 600, 700, 800, 900 and 1000 cycles at 1 C. Capacity changing during 1000 cycles at 1 C is demonstrated in Fig. 2 and capacity retention rates at 0.2 C are shown in Table 1. | ||
| Fig. 2 Capacity change of the LiCoO2/graphite battery during cycling at 1 C. | ||
| Cycle number | Capacity retention rates at 0.2 C (%) |
|---|---|
| 600 | 86.96 |
| 700 | 82.66 |
| 800 | 74.80 |
| 900 | 76.71 |
| 1000 | 71.69 |
The capacity of the LiCoO2/graphite battery at 0.2 C decreased with increased cycling. Moreover, the capacity declined clearly after the 800th cycle. This result indicated that obvious changes must have taken place inside the battery from the 800th cycle.
Fig. 3 shows SEM images of graphite electrodes after different numbers of cycles. The surfaces of pristine and activated electrodes were uniform, but some substances emerged after 600 and 700 cycles. The substances were fairly thick after 800 and 900 cycles, and covered a large proportion of the electrode after 1000 cycles. Therefore, the growth of these substances with cycling was consistent with the change in capacity. So the capacity loss of the battery may be related to these substances on the graphite surface. In addition, the substances were distributed unevenly on the surfaces of the cycled electrodes.
 | ||
| Fig. 3 SEM images of graphite electrodes after different cycles: (a) pristine; (b) activated; (c) 600 cycles; (d) 700 cycles; (e) 800 cycles; (f) 900 cycles; and (g) 1000 cycles. | ||
It could be seen from Fig. 4a that pristine graphite was composed of graphite layers with a clean surface. After 600 and 1000 cycles (Fig. 4c and f), two regions appeared on the surfaces of the electrodes, one without deposition and the other with deposition. A membrane was formed on the surface of cycled graphite in the region without deposition (Fig. 4d and g), which could be postulated as being SEI film.14–16 The deposition was composed of dendritic and grainy material (Fig. 4b and e). The morphology of deposition was the same as that of lithium deposition in lithium batteries17,18 and overcharged lithium ion batteries.19,20 The morphologies of the depositions on the electrodes after 600 and 1000 cycles were almost the same, but the deposition layer on the electrode cycled 1000 times was thicker than that cycled 600 times (Fig. 4c and f).
 | ||
| Fig. 4 SEM images of different regions on the surface of the graphite electrodes after different cycles: (a) pristine; (b–d) 600 cycles; (e–g) 1000 cycles. | ||
The surface morphology of cycled graphite and lithium deposits was examined by AFM. The surface of cycled graphite (Fig. 5a and c) was not flat, but large planes still existed. the rugged deposition (Fig. 5d) was composed of many protuberances with particle size of 0.7–2.2 μm (Fig. 5b and d). The deposits grew non-uniformly on the surface of the graphite anode.
 | ||
| Fig. 5 AFM images of cycled graphite and lithium deposits. (a) Two-dimensional height image of cycled graphite. (b) Two-dimensional height image of lithium deposits. (c) Three-dimensional height image of cycled graphite. (d) Three-dimensional height image of lithium deposits. | ||
To verify the existence of metallic lithium on the graphite anode after long-term cycles, DSC analysis was conducted on the cycled graphite anode with the deposition at the fully charged state. An endothermic peak at 180 °C appeared in the DSC curve in Fig. 6 and it corresponded to the melting of metallic lithium.21,22 Accordingly, metallic lithium surely existed in the deposition on the surface of long-term cycled graphite anode at charged state.
 | ||
| Fig. 6 DSC curve of the cycled graphite anode at fully charged state. | ||
In order to examine the surface compositions of the regions with and without deposition, XPS tests were performed on the two regions of the graphite anode after 1000 cycles. XPS curves and fitting results of Li 1s, C 1s, O 1s and F 1s are shown in Fig. 7. Peaks at 56.2 eV and 54.2 eV in Li 1s spectra could be attributed to LiF23,24 and Li2O,25,26 respectively. The peak at 55.3 eV was assigned to Li2CO3,27,28 LiOH29,30 and ROCO2Li.29 In the C 1s spectra, peaks of C–C at 284.6 eV and C–H at 285.2 eV were from ROCO2Li, (–CH2CH2O–)n (PEO) and ROLi. Peaks of C–O at 286.5 eV and CO32− at 289 eV came from PEO, ROLi, ROCO2Li and Li2CO3. Peaks of Li–O–Li at 529.3 eV, Li–O–H and CO32− at 531.9 eV, and C–O at 533.5 eV appeared in O 1s spectra, which proved the formation of Li2O, LiOH, ROCO2Li, Li2CO3, PEO and ROLi.31 Only a peak of LiF at 686.2 eV32 was observed in the F 1s spectra.
 | ||
| Fig. 7 XPS curves and fitting results of Li 1s, C 1s, O 1s and F 1s in the regions with and without depositions on the surface of graphite electrodes after 1000 cycles. (a), (c), (e) and (g): the region with depositions. (b), (d), (f) and (h): the region without depositions. | ||
LiF, Li2O, Li2CO3, LiOH, ROCO2Li, ROLi and PEO appeared on the surfaces of both cycled graphite and deposition as seen from the above XPS results. Therefore the SEI films were formed on the surfaces of both cycled graphite and lithium deposits. Peak positions in fitted Li 1s, C 1s, O 1s and F 1s spectra of the two regions were the same, which demonstrated that the composition of the SEI film on the surface of the lithium deposits was the same as that on the surface of cycled graphite.
The battery was dismantled after certain cycles and fresh electrolyte was added into the battery. The effect of cycled electrolyte on the capacity loss of the battery was examined by charging and discharging at 0.2 C. Charge/discharge curves of pristine battery, cycled battery and the cycled battery with fresh electrolyte added are shown in Fig. 8. The shapes of the three charge/discharge curves were nearly the same. Capacity changes at 0.2 C are shown in Table 2. The capacity retention rate of the cycled battery was 82.62%, which meant capacity loss was 17.38%. The capacity appeared to show a slight recovery after the addition of fresh electrolyte into the cycled battery. The capacity retention rate returned to 86.72%, which demonstrated that the effect of battery components other than the electrolyte on capacity loss was 13.28%. If the total capacity loss of 179.6 mA h after the cycling was defined as 100%, the capacity loss of 42.4 mA h (179.6 − 137.2 = 42.4) from the electrolyte was 23.61% and the capacity loss from the other components was 76.39%.
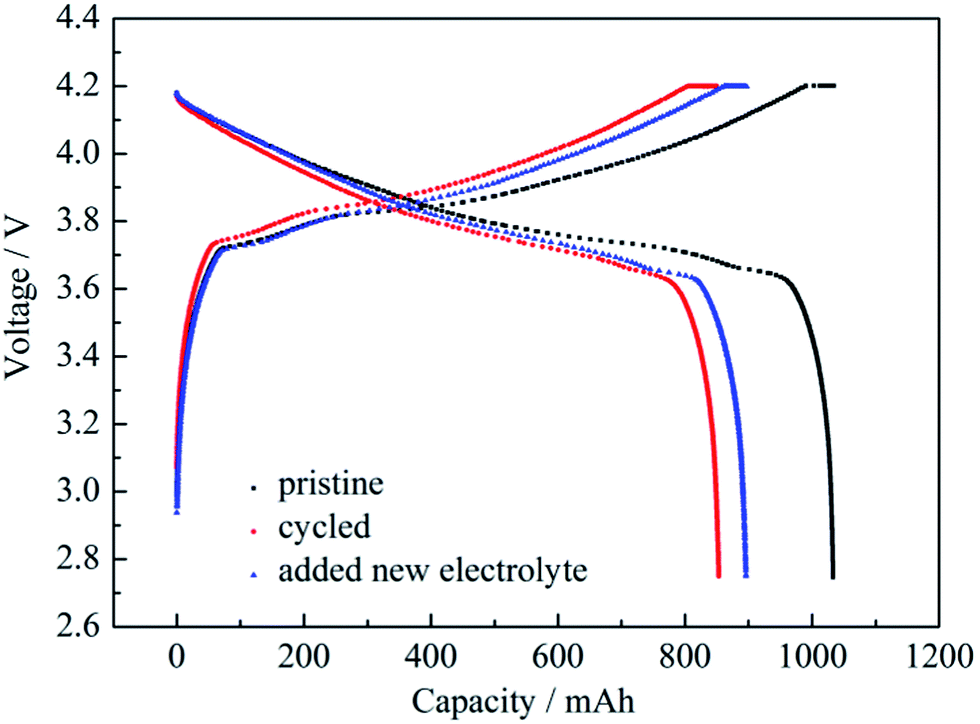 | ||
| Fig. 8 Charge/discharge curves at 0.2 C of pristine battery, cycled battery and the cycled battery with fresh electrolyte added. | ||
| Capacity at 0.2 C (mA h) | Capacity retention rates at 0.2 C (%) | Capacity loss (mA h) | |
|---|---|---|---|
| Pristine battery | 1033.1 | 100.00 | 0 |
| Cycled battery | 853.5 | 82.62 | 179.6 |
| The cycled battery with fresh electrolyte added | 895.5 | 86.72 | 137.2 |
From the XPS results, SEI film appeared on the surface of the lithium deposits. The SEI film was composed of reaction products of lithium deposits reacting with the electrolyte. Therefore, the appearance of lithium deposits led to consumption of the electrolyte. In addition, the formation of SEI film on the surfaces of graphite and LiCoO2 electrodes could also consume electrolyte in the battery. These consumptions of electrolyte would decrease the active lithium and ionic conductivity of the battery, which was one of the reasons for the capacity loss of the battery.
Lithium deposition itself also consumed the battery's active lithium. More deposits appeared on the anode with the cycling, so more active lithium was consumed, also leading to capacity loss of the battery.
The electrode potential of the graphite anode during the charging and discharging of a full battery was examined by a three-electrode cell. Such experiments were conducted on two batteries after different numbers of cycles. The capacity retention rate of battery #1 was 97.78%, and that of #2 was 82.62%. These two batteries were charged and discharged at 1 C. Voltage–time curves of the full batteries and their corresponding graphite anodes are shown in Fig. 9.
 | ||
| Fig. 9 Voltage–time curves of full batteries #1, #2 and corresponding graphite anodes during the charging and discharging of the two batteries at 1 C. | ||
The charge/discharge voltage ranges of graphite anodes were within the range of 0.09–0.19 V. For the charging process of full battery, anode potential decreased continuously during constant-current charging, and then increased slightly during the following constant-voltage charging. Therefore the lowest anode potential appeared at the end of constant-current charging of full battery. The lowest anode potential of #1 was 0.103 V and that of #2 was 0.096 V, which indicated that the lowest anode potential of #2 was more negative than that of #1. The anode potential measured by the three-electrode cell was the average potential of the whole graphite anode. Maybe the local potential of the cycled anode was below 0 V (vs. Li+/Li), leading to the deposition of lithium metal. Because the lithium deposits in #2 were more than those in #1, the average potential of #2 at the end of constant-current charging was more negative than that of #1.
Conclusions
Lithium deposition was evaluated using LiCoO2/graphite batteries during long-term cycles. The batteries were charged and discharged at a rate of 1 C within the voltage range of 2.75–4.2 V for 600, 700, 800, 900 and 1000 cycles. Capacity obviously declined from the 800th cycle, which was consistent with the change of surface morphology of the graphite anode. As the cycling proceeded, more lithium deposits were also formed on the surface of the graphite besides the SEI film. The dendritic and granular deposits were distributed non-uniformly. DSC results verified the existence of metallic lithium in the deposits. SEI film was also formed on the surface of the lithium deposits. The components were the same as those of the SEI film on the surface of graphite, which were LiF, Li2O, Li2CO3, LiOH, ROCO2Li, ROLi and PEO. Formation of these surface films would consume the electrolyte in the battery. Capacity loss from the electrolyte was 23.61%, while capacity loss from other battery components was 76.39%. In addition, the formation of lithium deposits also consumed active lithium and caused capacity loss. Local potential of the graphite anode below 0 V led to the generation of lithium deposits, making the average anode potential at the end of constant-current charging for the full battery more negative during the cycling.Acknowledgements
The authors would like to thank the National High Technology Research and Development Program (863 Program) of China (no. 2012AA110203) for financial support.Notes and references
- Y. Takanashia, Y. Orikasaa, M. Mogia, M. Oishia, H. Murayamaa, K. Satoa, H. Yamashigea, D. Takamatsua, T. Fujimotoa, H. Tanidaa, H. Araia, T. Ohtab, E. Matsubarac, Y. Uchimotod and Z. Ogumia, J. Power Sources, 2011, 196, 10679 CrossRef PubMed
.
- M. Jo, Y.-S. Hong, J. Choo and J. Cho, J. Electrochem. Soc., 2009, 156, A430 CrossRef CAS PubMed
- J. T. Lee, N. Nitta, J. Benson, A. Magasinski, T. F. Fuller and G. Yushin, Carbon, 2013, 52, 388 CrossRef CAS PubMed
- M. Lu, H. Cheng and Y. Yang, Electrochim. Acta, 2008, 53, 3539 CrossRef CAS PubMed
- X. Wang, Y. Sakiyama, Y. Takahashi, C. Yamada, H. Naito, G. Segami, T. Hironaka, E. Hayashi and K. Kibe, J. Power Sources, 2007, 167, 162 CrossRef CAS PubMed
- H. P. Zhang, L. J. Fu, Y. P. Wu and H. Q. Wu, Electrochem. Solid-State Lett., 2007, 10, A283 CrossRef CAS PubMed
- C.-N. Li, J.-M. Yang, V. Krasnov, J. Arias and K.-W. Nieh, Electrochem. Solid-State Lett., 2008, 11, A81 CrossRef CAS PubMed
- P. L. Moss, G. Au, E. J. Plichta and J. P. Zhenga, J. Electrochem. Soc., 2010, 157, A1 CrossRef CAS PubMed
- S. S. Zhang, K. Xu and T. R. Jow, J. Power Sources, 2006, 160, 1349 CrossRef CAS PubMed
- M.-S. Wu, P.-C. J. Chiang and J.-C. Lin, J. Electrochem. Soc., 2005, 152, A47 CrossRef CAS PubMed
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J.-G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450 CrossRef CAS PubMed
- G. Park, N. Gunawardhana, H. Nakamura, Y.-S. Lee and M. Yoshio, J. Power Sources, 2012, 199, 293 CrossRef CAS PubMed
- P. Arora, M. Doyle and R. E. White, J. Electrochem. Soc., 1999, 146, 3543 CrossRef CAS PubMed
- H. Buqa, A. Würsig, J. Vetter, M. E. Spahr, F. Krumeich and P. Novák, J. Power Sources, 2006, 153, 385 CrossRef CAS PubMed
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332 CrossRef CAS PubMed
- M. Kassem and C. Delacourt, J. Power Sources, 2013, 235, 159 CrossRef CAS PubMed
- K. Kanamura, H. Tamura, S. Shiraishi and Z. Takehara, J. Electroanal. Chem., 1995, 394, 49 CrossRef
- C. M. López, J. T. Vaughey and D. W. Dees, J. Electrochem. Soc., 2012, 159, A873 CrossRef PubMed
- W. Lu, C. M. López, N. Liu, J. T. Vaughey, A. Jansen and D. W. Dees, J. Electrochem. Soc., 2012, 159, A566 CrossRef CAS PubMed
- D. Belov and M.-H. Yang, J. Solid State Electrochem., 2008, 12, 885 CrossRef CAS
- H. Maleki, G. Deng, A. Anani and J. Howard, J. Electrochem. Soc., 1999, 146, 3224 CrossRef CAS PubMed
- T. Kawamura, A. Kimura, M. Egashira, S. Okada and J.-I. Yamaki, J. Power Sources, 2002, 104, 260 CrossRef CAS
- K. Kanamura, H. Tamura, S. Shiraishi and Z.-I. Takehara, J. Electroanal. Chem., 1995, 394, 49 CrossRef
- D. Aurbach, I. Weissman, A. Schechter and H. Cohen, Langmuir, 1996, 12, 3991 CrossRef CAS
- K. Kanamura, S. Shiraishi, H. Tamura and Z.-I. Takehara, J. Electrochem. Soc., 1994, 141, 2379 CrossRef CAS PubMed
- K. Edström, M. Herstedt and D. P. Abraham, J. Power Sources, 2006, 153, 380 CrossRef PubMed
- K.-I. Morigaki and A. Ohta, J. Power Sources, 1998, 76, 159 CrossRef CAS
- A. M. Andersson, A. Henningson, H. Siegbahn, U. Jansson and K. Edström, J. Power Sources, 2003, 119–121, 522 CrossRef CAS
- D. Bar-Tow, E. Peled and L. Burstein, J. Electrochem. Soc., 1999, 146, 824 CrossRef CAS PubMed
- K. Kanamura, H. Tamura and Z. Takehara, J. Electroanal. Chem., 1992, 333, 127 CrossRef CAS
- L. Yang, X. Cheng, Y. Ma, S. Lou, Y. Cui, T. Guan and G. Yin, J. Electrochem. Soc., 2013, 160, A2093 CrossRef CAS PubMed
- V. Eshkenazi, E. Peled, L. Burstein and D. Golodnitsky, Solid State Ionics, 2004, 170, 83 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2014 |