A new recyclable 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2 ionic liquid reagent for selective bromination of anilines or phenols and α-bromination of alkanones under mild conditions†
Hojat Veisi*a,
Alireza Sedrpoushan*b,
Pourya Mohammadia,
Ali Reza Farajic and
Sami Sajjadifara
aDepartment of Chemistry, Payame Noor University(PNU), 19395-4697 Tehran, Iran. E-mail: hojatveisi@yahoo.com; Fax: +98 831 8380709
bInstitute of Industrial Chemistry, Iranian research Organization for Science and Technology, Tehran, Iran
cFaculty of chemistry, Pharmaceutical Sciences Branch, Islamic Azad University, Tehran, Iran
First published on 3rd June 2014
Abstract
1,4-Bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2 has been synthesized and explored as a new efficient brominating agent. The crystalline ditribromide reagent is stable for months and acts as a safe source of bromine requiring just 0.5 equiv. for complete bromination. It has a high active bromine content per molecule and shows a remarkable reactivity toward various substrates in acetonitrile at room temperature. The prepared reagents were used as a green recyclable reaction media for the selective bromination of anilines, phenols and α-bromination of alkanones in excellent yields. The product can easily be isolated by just washing the highly water soluble 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2 from the brominated product. The spent reagent can be recovered, regenerated, and reused without any significant loss.
Bromination of aromatic compounds and α-bromination of alkanones have attracted much attention in recent years1 for the synthesis of biologically active compounds such as potent antitumor, antibacterial, antifungal, antiviral, and antioxidizing agents. Numerous industrially valuable products such as pesticides, insecticides, herbicides, fire retardants, and other new materials carry the bromo functionality.2 These halides also undergo carbon–carbon bond formation3–5 or carbon–heteroatom bond formation via aromatic functionalization protocols.6
Organic tribromide reagents (OTBs) are preferable as oxidants to molecular bromine, owing to the hazards associated with elemental bromine. Several tribromides have been reported, that is, tetramethylammonium tribromide,7 phenyltrimethylammonium tribromide,8 cetyltrimethylammonium tribromide, tetrabutylammonium tribromide,9 1,8-diazabicyclo[5,4,0]-tetrabutylammonium tribromide,10 1,2-dipyridiniumditribromide-ethane (DPTBE),11 pyridine hydrobromide perbromide,12 hexamethylenetetramine-bromine,13 and DABCO–bromine.14 {[K.18-crown-6]Br3}n,15 1,3-di-n-butylimidazolium tribromide [BBIm]Br3,16 ethylenebis(N-methylimidazolium) ditribromide (EBMIDTB),17 1-butyl-3-methylpyridinium tribromide ([BMPy]Br3)18 and BBIm]Br3.19 Recently some ionic liquid tribromides (IL-Br3−) for the preparation of bromoesters from aromatic aldehydes20 and bromination of aromatic substrates21 were reported. It would be extremely useful to develop further synthetic protocols for the synthesis of organic tribromide reagents.22
Development of environmentally benign and efficient processes with simple work-up and high purity of the products with high yields is currently receiving considerable attention. In this context, tribromide ionic liquid plays an important role in the process of obtaining monobromo derivatives with the ultimate goal of hazard-free, waste-free and solvent-free efficient synthesis of biologically active compounds that can be used as precursors of natural products.23,24
In recent years, there has been considerable interest in developing green chemistry25 for organic synthesis due to environmental demand and sustainability. There are various kinds of alkyl imidazolium salts which are commercially available as room temperature ionic liquids (RTILs). The combination of alkylimidazolium cation with the tribromide anion should therefore lead to RTIL bromine analog. Ionic liquids were introduced as alternative green reaction media because of their unique chemical and physical properties of non-volatility, noninflammability, thermal stability, and ease of recyclability.
Tribromides are more suitable than the liquid bromine because of their crystalline nature, hence easy for their storage, transport, and maintenance of desired stoichiometry. Preparations of these reagents involve organic ammonium bromide and molecular bromine in most cases, thus an indirect use of toxic molecular bromine. So, in continuation of our interest in organohalogen chemistry,26 herein, we have synthesized a novel ditribromide reagent. The new reagent has higher bromine content per molecule, better bromination efficiency and selectivity and is devoid of phase transfer property, and the spent reagent can be recovered and regenerated easily. In this paper we wished to report the preparation of a new ditribromide reagent (Scheme 1), development of a metal and ionic-liquid free bromination protocol, and recovery of the reagent. The reagent was prepared by treating 1-methylimidazole (2 equiv.) with 1,4-dibromobutane (1 equiv.) at room temperature. The resultant 1,4-bis(3-methylimidazolium-1-yl)butane di(bromide) [bMImB]·(Br)2 as light brown crystals was treated with molecular bromine. The orange precipitate of 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2 was filtered (96% yield) and was recrystallized in acetonitrile to obtain large crystals as shown in Fig. 1. The compound has been characterized by spectral and analytical data. This crystalline compound (Fig. 1) is stable for several months at room temperature without loss of its activity.
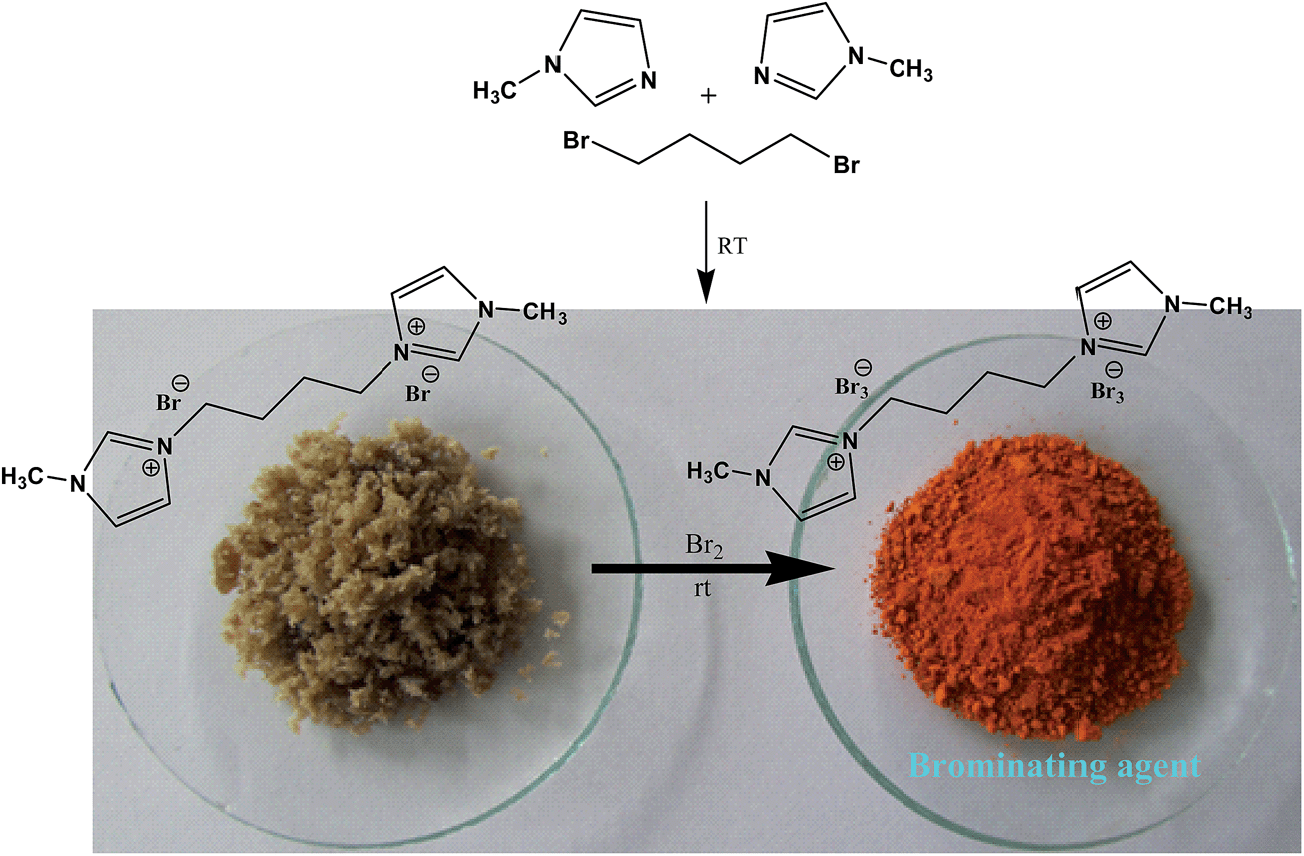 | ||
| Scheme 1 | ||
 | ||
| Fig. 1 Crystals of 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2. | ||
The [bMImB]·(Br3)2 showed an intense UV absorption at 267 nm (Fig. 2), typical for tribromide ion (Br3−).27
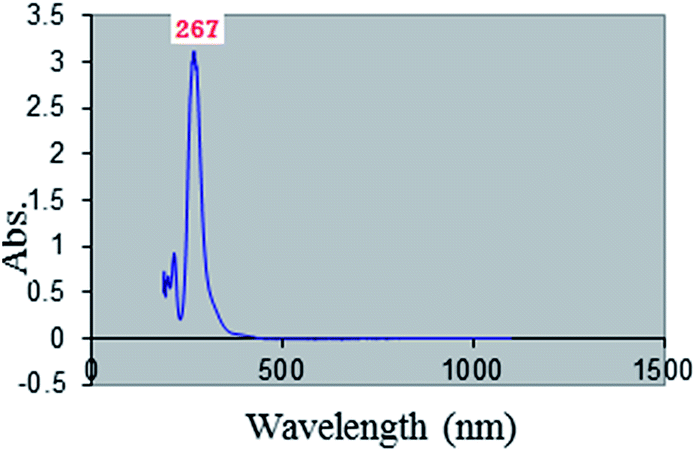 | ||
| Fig. 2 The UV absorption of [bMImB]·(Br3)2. | ||
[bMImB]·(Br3)2 was used as an alternative brominating agent for bromination of activated aromatic compounds and α-bromination of alkanones (Scheme 2). It is worthy to be noted that bromination of organic compounds is usually effected by using equimolar ratios of bromine or tribromide with the substrate. To test the bromination efficiency of this new reagent, 0.5 equiv. of the reagent 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2 was added to 1.0 equiv. of phenol in acetonitrile (3 mL). The reaction was completed within 10 minutes in 96% yield. Thus, [bMImB]·(Br3)2 efficiently delivers the bromine to the reaction medium and itself gets recovered quantitatively with adding water, thereby acting as the perfect bromine carrier.
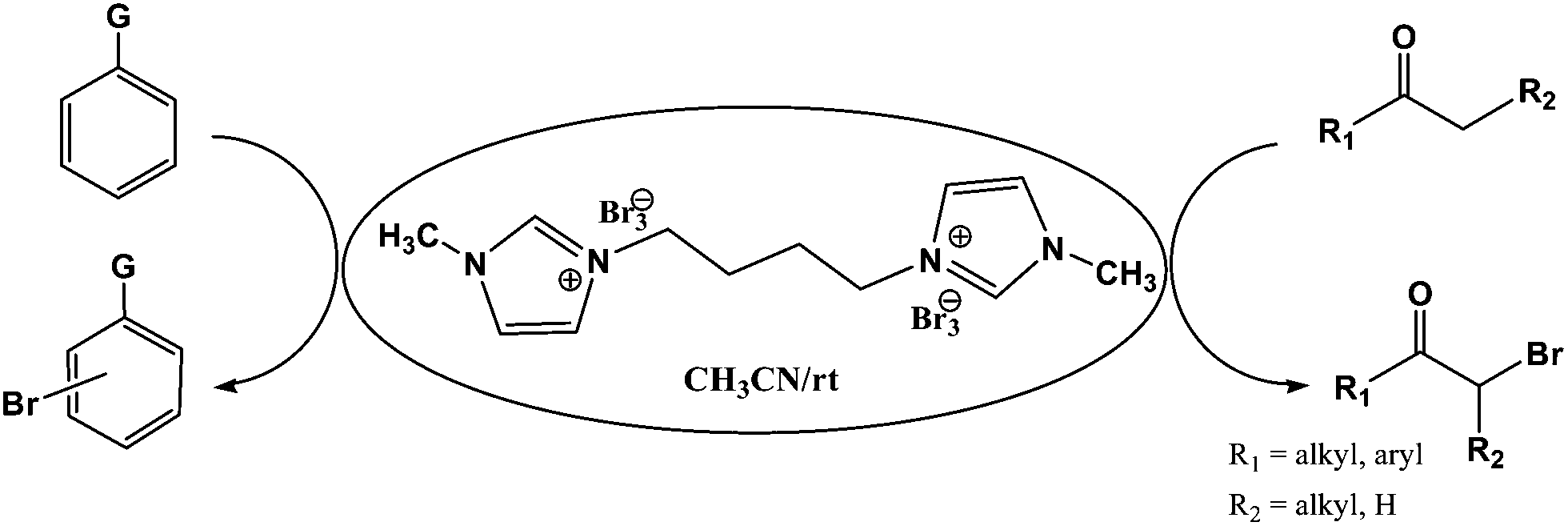 | ||
| Scheme 2 | ||
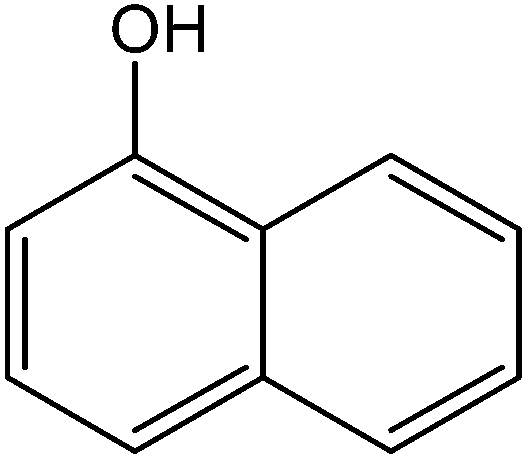
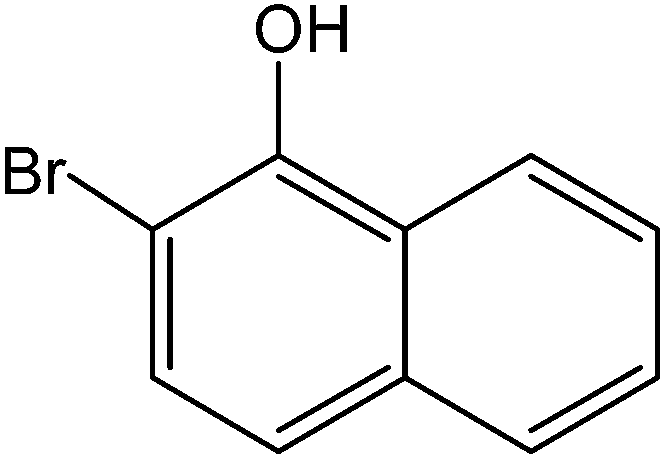
Various anilines and phenols were monobrominated with complete selectivity in excellent yields at room temperature, and the results are summarized in Table 1. Phenol and aniline were cleanly monobrominated, affording exclusively p-bromophenol and p-bromoaniline (entries 1, 2). Regioselective bromination of phenol and aniline is an important protocol in organic synthesis because of their versatile synthetic intermediates for a considerable number of useful transformations. In an aromatic electrophilic substitution reaction para-orientation is favored over ortho-orientation due to stereoelectronic effects. Similarly, a variety of substituted anilines and pheols underwent regioselective monobromination (entries 3–6). α-Naphthol and β-naphthol, being a bulky molecule, took 40–50 min for monobromination (entries 7, 8).
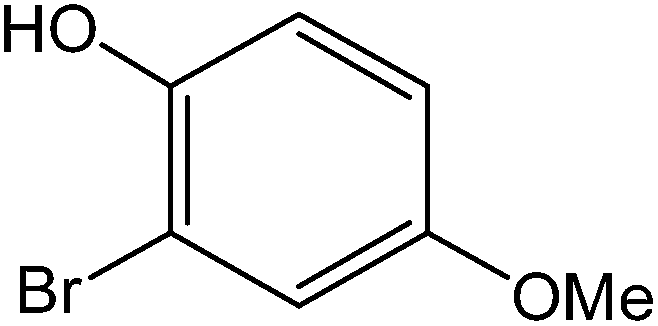
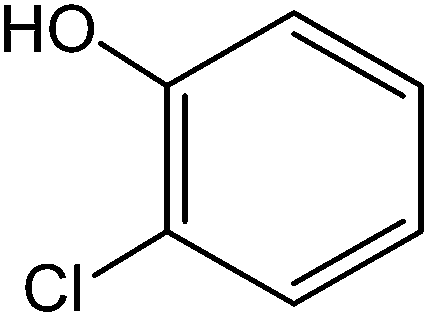
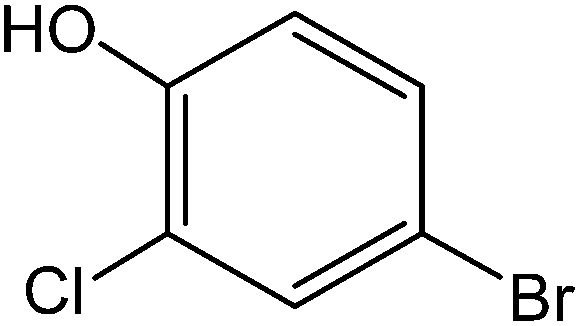
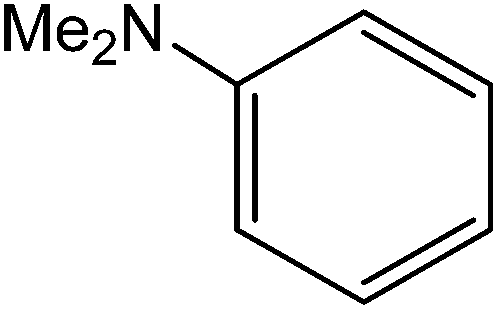

All the reactions were quenched by adding water. This caused precipitation of the products as solids or oils, which were readily extracted with ethyl acetate and then dried (either with sodium sulfate or in vacuo). The aqueous phase containing highly water-soluble IL was easily concentrated in vacuo to recycle [bMImB]·(Br)2, which then could be used to regenerate [bMImB]·(Br3)2 with bromine (Scheme 3). Regeneration and reusability are two significant features of ionic liquids.28 The effectiveness of ionic liquid, their recyclability and regeneration processes were observed for the bromination of phenol and found to be effective up to 5 cycles (Fig. 3).
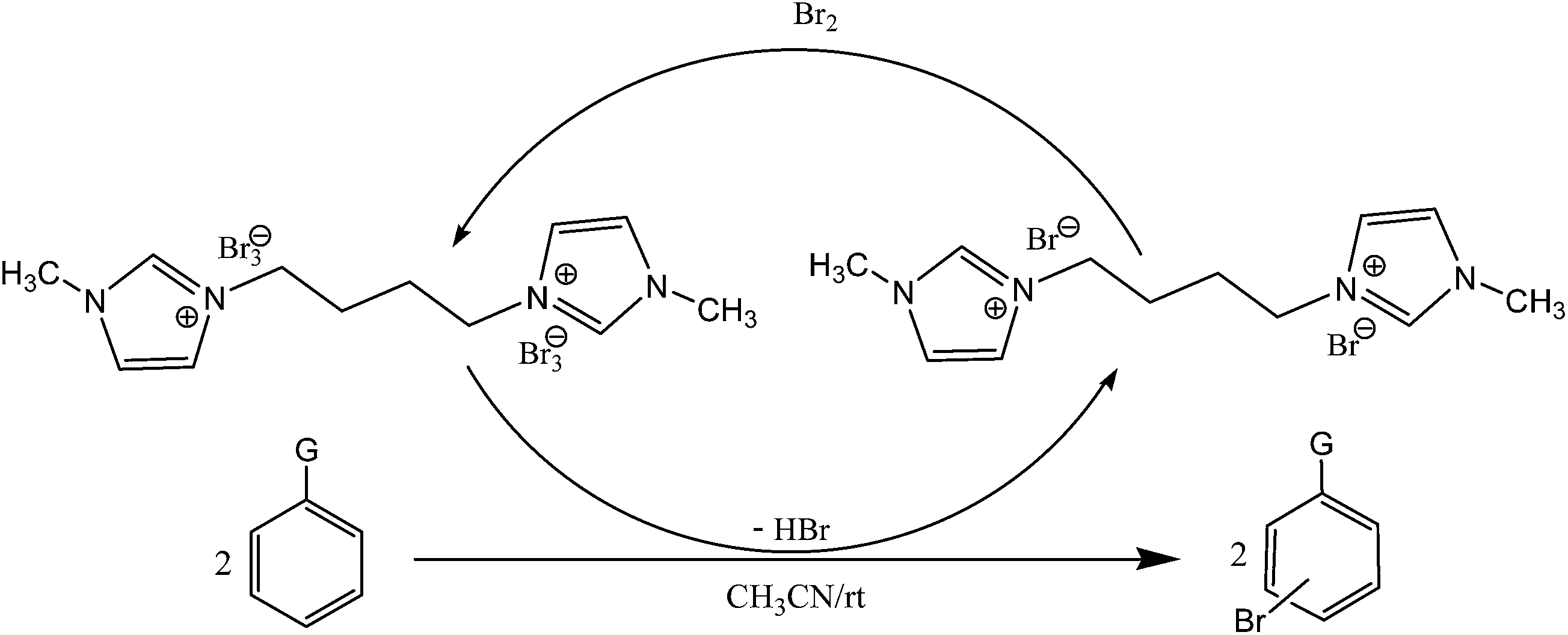 | ||
| Scheme 3 | ||
 | ||
| Fig. 3 Reusability of ionic liquid [bMImB]·(Br3)2 in the bromination of phenol. | ||
The plausible mechanism is shown in Scheme 4. [bMImB]·(Br3)2 exists in a rapid equilibrium with imidazolium bromide and bromine in solution.
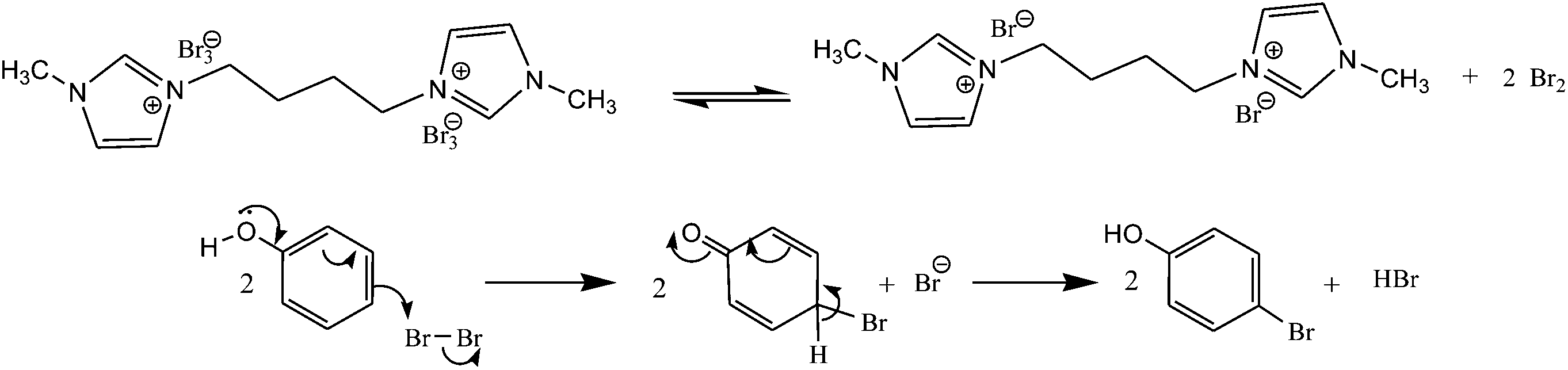 | ||
| Scheme 4 Plausible mechanism. | ||
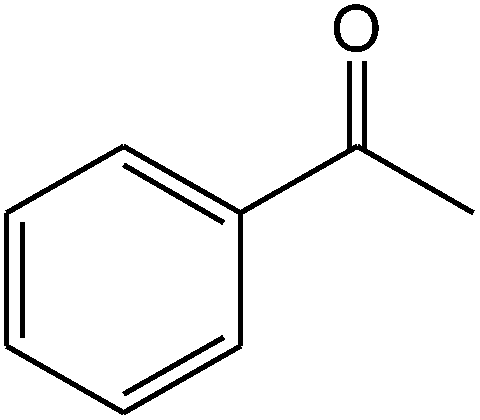
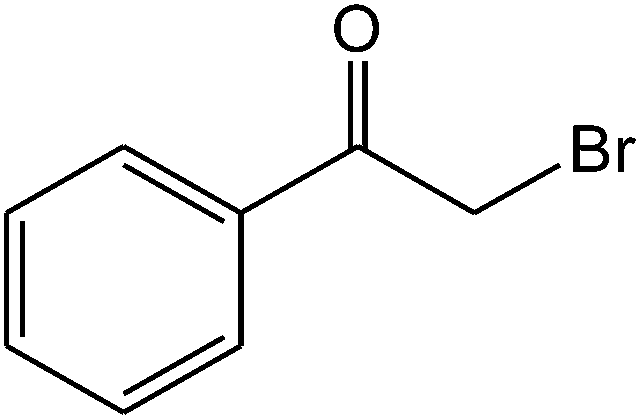
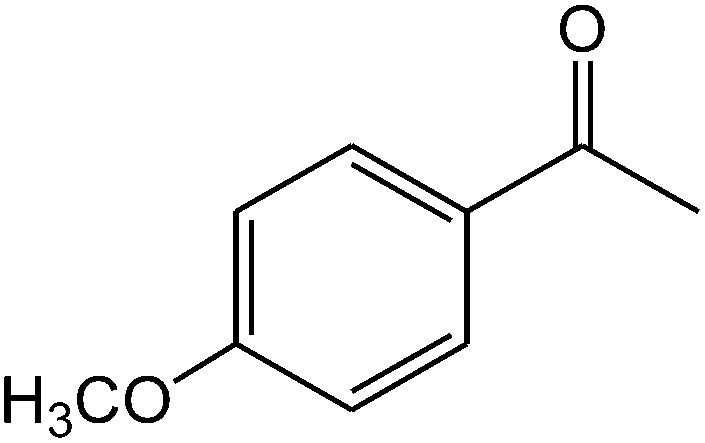
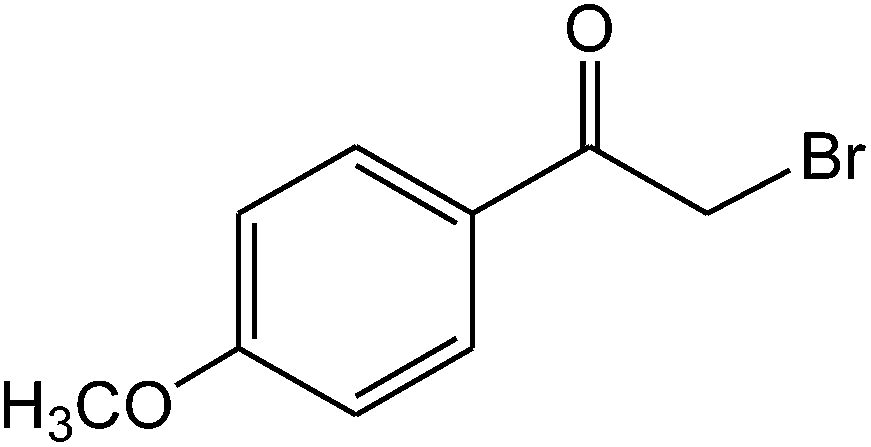
The ionic liquid under investigation was also effective for the selective bromination of alkanones (Scheme 2). We found that the reaction of acetophenone with [bMImB]·(Br3)2, could occur rapidly in acetonitrile at room temperature and completed within 30 min to give α-bromoacetophenone (Table 2, entry 1). In similar fashion, the reaction of [bMImB]·(Br3)2 with a variety of alkanones was investigated, we found that the reaction is general and applicable to several substituted acetophenone containing different substitutes, such as methyl and methoxy (Table 2, entries 2,3). In order to explore the generality of the method developed for the synthesis of monobromination of alkanones, we conducted the experiments with [bMImB]·(Br3)2 to ethyl 3-oxo-3-phenylpropanoate and ethyl acetoacetate, which were also effective and gave the corresponding monobromination products in excellent yields (Table 2, entries 4, 5). To investigate the selectivity of this reagent, ethyl methyl ketone as dissymmetric dialkyl ketone was chosen, but we could not find any selectivity under this system.


In conclusion, we have reported a novel ditribromide ionic liquid reagent with high active bromine content per molecule, which acts as a good brominating agent for brominating 1 equiv. of the substrate with just 0.5 equiv. of the reagent. The prepared reagent were used as a green recyclable reaction media for the selective bromination of anilines, phenols and α-bromination of alkanones in excellent yields under mild conditions. The significant characteristics of the method which are worth mentioning include high conversions, short process time, mild reaction conditions, environmentally friendly process, simple workup with good to quantitative yields and re-usable ionic liquid.
Experimental section
Preparation of 1,4-bis(3-methylimidazolium-1-yl)butane dibromide [bMImB]·(Br)2
1,4-Dibromobutane (10.79 g, 50.0 mmol) was mixed with 1-methylimidazole (8.16 g, 99.5 mmol) and the reaction mixture was kept at room temperature for 48 h. The resulting dark solid was crushed under acetone, filtered, washed with small portions of acetone and dried under reduced pressure, leaving the desired 1,4-bis(3-methylimidazolium-1-yl)butane di(bromide) [bMImB]·(Br)2 as light brown crystals (14.62 g, 77% yield). Melting point: 126–128 °C. IR (KBr) 3146, 3093, 3088, 2966, 2856, 1635, 1575, 1456, 1427, 1168, 856, 788, 758, 624 cm−1. 1H NMR (D2O) (ppm) d: 8.59 (s, 2H, CH imidazolium); 7.33 (t, 2H, CH imidazolium); 7.29 (s, 2H, CH imidazolium); 4.10 (t, 4H, NCH2); 3.74 (s, 6H, NCH3); 1.76 (m, 4H, CH2CH2). 13C NMR (CDCl3) (ppm) d: 136.8, 129.1 and 118.6 (CH imidazolium); 46.6 (NCH2); 30.6 (NCH3); 25.8 (CH2).Preparation of 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2
In a fume cupboard, molecular bromine (1.0 mL, 2.0 mmol) was added dropwise over 10 min to 1,4-bis(3-methylimidazolium-1-yl)butane dibromide (0.380 g, 1.0 mmol) under stirring and cooling in an ice-bath affording a deep orange solid [bMImB]·(Br3)2 ionic liquid, and stirring was continued for 2 h. The orange precipitate was recrystallized from acetonitrile to yield 1.77 g (96% yield) of 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2. Melting point: 132 °C. UV (CH3CN) 267 nm. IR (KBr) 3158, 3136, 3086, 2966, 2856, 1635, 1595, 1570, 1556, 1464, 1400, 1162, 1103, 837, 742, 617 cm−1.1H NMR (D2O) (ppm) d: 8.83 (s, 2H, CH imidazolium); 7.55 (m, 4H, CH imidazolium); 4.33 (t, 4H, NCH2); 3.96 (s, 6H, NCH3); 1.99 (m, 4H, CH2CH2). 13C NMR (CDCl3) (ppm) d: 123.7, 123.6 and 122.0 (CH imidazolium); 48.6 (NCH2); 35.8 (NCH3); 26.0 (CH2). Anal. calcd for C12H20Br6N4: C, 20.60; H, 2.88; N, 8.01; Br, 68.51. Found: C, 20.10; H, 2.19; N, 7.65; Br, 67.87%.General procedure for bromination of anilines or phenols
To a mixture of anilines or phenols (2.0 mmol) in 2 mL acetonitrile, was added 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide (1.0 mmol) and stirred at room temperature. After disappearance of the starting material (monitored by TLC; n-hexane–acetone 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), water was added into the reaction mixture to quench the IL. The resulting reaction mixture was extracted with ethyl acetate, separated from the organic layer, dried over sodium sulfate, and concentrated to afford pure monobromo anilines or monobromo phenols.
:1), water was added into the reaction mixture to quench the IL. The resulting reaction mixture was extracted with ethyl acetate, separated from the organic layer, dried over sodium sulfate, and concentrated to afford pure monobromo anilines or monobromo phenols.
General procedure for bromination of alkanones
To a mixture of alkanone (2.0 mmol) in 2 mL acetonitrile, was added 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide (1.0 mmol) and stirred at room temperature. After disappearance of the starting material (monitored by TLC; n-hexane–acetone 7:1), water was added into the reaction mixture to quench the IL. The resulting reaction mixture was extracted with diethyl ether, separated from the organic layer, dried over sodium sulfate, and purified by flash column chromatography over silica gel (hexane–EtOAc 10/2).
Regeneration of ionic liquid
After the addition of water into the reaction mixture, the water soluble ionic liquid 1,4-bis(3-methylimidazolium-1-yl)butane dibromide [bMImB]·(Br)2 was recovered by evaporation of the aqueous solution and washing with diethyl ether in each case. The recovered ionic liquid [bMImB]·(Br)2 was dried at 40 °C for 12 h, and treated with molecular bromine (2 mmol) under stirring and cooling in an ice-bath for 2 h to regenerate 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide [bMImB]·(Br3)2 ionic liquid and reused for the subsequent reaction without loss of activity.References
- R. C. Larock, Comprehensive Organic Transformations, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed
.
-
(a) G. W. Gribble, Chem. Soc. Rev., 1999, 28, 335 RSC
-
(a) J. K. Stille, Pure Appl. Chem., 1985, 57, 1771 CrossRef CAS
-
(a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed
-
(a) K. Sonogashira, Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol. 3, p. 521 Search PubMed
-
(a) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS
-
(a) F. D. Cattaway and G. Hoyle, J. Chem. Soc., 1923, 654 RSC
- S. Kajigneshi, T. Kakinami, H. Tokiyama, T. Hirakawa and T. Okamoto, Chem. Lett., 1987, 627 CrossRef
- R. E. Buckles, A. I. Popov, W. F. Zelezny and R. J. Smith, J. Am. Chem. Soc., 1951, 73, 4525 CrossRef CAS
- H. A. Muathen, J. Org. Chem., 1992, 57, 2740 CrossRef CAS
- V. Kavala, S. Naik and B. K. Patel, J. Org. Chem., 2005, 70, 4267 CrossRef CAS PubMed
- L. F. Fieser and M. Fieser, Reagents for Organic Synthesis, Wiley, 1967, p. 967 Search PubMed
- I. Yavari and A. Shaabani, J. Chem. Res. (S), 1994, 7, 274 Search PubMed
- M. M. Heravi, F. Derikvand, M. Ghassemzadeh and B. Neumuller, Tetrahedron Lett., 2005, 46, 6243 CrossRef CAS PubMed
- M. A. Zolfigol, G. Chehardoli, S. Salehzadeh, H. Adams and M. D. Word, Tetrahedron Lett., 2007, 48, 7969 CrossRef CAS PubMed
- S. P. Borikar and T. Daniel, J. Iran. Chem. Soc., 2011, 8, 531 CrossRef CAS
- R. Hosseinzadeh, M. Tajbakhsh, M. Mohadjerani and Z. Lasemi, Synth. Commun., 2010, 40, 868 CrossRef CAS
- S. P. Borikar, T. Daniel and V. Paul, Tetrahedron Lett., 2009, 50, 1007 CrossRef CAS PubMed
- S. P. Borikar, T. Daniel and V. Paul, Synth. Commun., 2010, 40, 647 CrossRef CAS
- W. Bao and Z. Wang, Green Chem., 2006, 8, 1028 RSC
- C. Chiappe, E. Leandri and D. Pieraccini, Chem. Commun., 2004, 253 Search PubMed
- M. K. Chaudhuri, A. T. Khan and B. K. Patel, Tetrahedron Lett., 1998, 39, 8163 CrossRef CAS
- P.-M. Léo, C. Morin and C. Philouze, Org. Lett., 2002, 4, 2711 CrossRef PubMed
-
(a) K. Tatsuta, H. Ozeki, M. Yamaguchi, M. Tanaka and T. Okui, Tetrahedron Lett., 1990, 31, 5495 CrossRef CAS
- I. T. Horvath and P. T. Anastas, Chem. Rev., 2007, 107, 2169 CrossRef CAS PubMed
- H. Veisi and R. Ghorbani-Vaghei, Tetrahedron, 2010, 66, 7445 CrossRef CAS PubMed
- M. K. Chaudhuri, A. T. Khan, B. K. Patel, D. Dey, W. Kharmawophlang, T. R. Lakshmiparbha and G. C. Mandal, Tetrahedron Lett., 1998, 39, 8163 CrossRef CAS
- M. P. Kaushik and V. Polshettiwar, Indian J. Chem., 2006, 45B, 2542 CAS
- S. J. Zhang and Z. G. Le, Chin. Chem. Lett., 2005, 16, 1590 CAS
- Z. G. Le, Z. C. Chen, Y. Hu and Q. G. Zheng, Chin. Chem. Lett., 2005, 16, 1007 CAS
- S. A. Pourmousavi and P. Salehi, Acta Chim. Slov., 2009, 56, 734 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03006k |
| This journal is © The Royal Society of Chemistry 2014 |