 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the mechanism of the Povarov reaction. A DFT study†
Luis R. Domingo*a,
María J. Aurella,
José A. Sáezb and
Sidi M. Mekellechec
aDepartamento de Química Orgánica, Universidad de Valencia, Dr Moliner 50, 46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es; Web: www.luisrdomingo.com
bInstituto de Tecnología Química UPV-CSIC, Camino de Vera s/n, 46022 Valencia, Spain
cLaboratory of Applied Thermodynamics and Molecular Modeling Department of Chemistry, Faculty of Science, University A. Belkaid of Tlemcen, PB 119, 13000, Algeria
First published on 29th May 2014
Abstract
The molecular mechanism of the Povarov reaction in acetonitrile has been studied at the MPWB1K/6-311G** level of theory. This reaction follows a domino process that comprises two sequential reactions: (i) a Lewis acid catalysed aza-Diels–Alder (A-DA) reaction between a N-aryl imine and a nucleophilic ethylene yielding a formal [4 + 2] cycloadduct; (ii) a stepwise 1,3-hydrogen shift at this intermediate affording the final tetrahydroquinoline. At this computational level, the Lewis acid catalysed A-DA reaction presents a two-step mechanism as a consequence of the large stabilisation of the corresponding zwitterionic intermediate. Our study allows establishing that the N-aryl substituent has no remarkable incidence in the activation energy, but the presence of a second C-aryl substituent has a relevant role in the reaction rate. Analysis of the DFT-based reactivity indices of the reagents provides further explanation of the behaviours of the mechanism of the A-DA reaction involved in the Povarov reaction.
Introduction
In its original form, the Povarov reaction1 (PR) involved a Lewis acid (LA) catalysed aza-Diels–Alder (A-DA) reaction between a N-aryl imine 1 (a formal 2-azadiene) and a vinyl ether 2 (an electron-rich (ER) ethylene), followed by a 1,3-hydrogen shift in the formal [4 + 2] cycloadduct 3 to yield a 1,2,3,4-tetrahydroquinoline 4 (Scheme 1).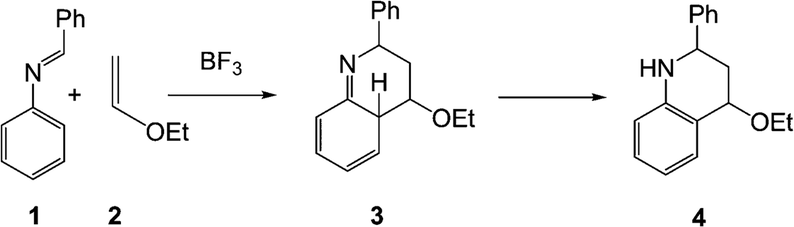 | ||
| Scheme 1 The Povarov reaction. | ||
Despite its impressive synthetic potential (two new C–C bonds, one new ring and up to three new stereogenic centers are formed), the PR received only sporadic attention from the time of its discovery (1960s) until the 1990s, when an one-pot, three-component version involving in situ generation of the N-aryl imine 1 by a condensation of a carbonyl compound with aniline, was developed (see Scheme 2).2 Either considered as a two-step sequence or as a multicomponent reaction, there are three points of diversity, which render the PR well-suited to diversity-oriented synthesis.3
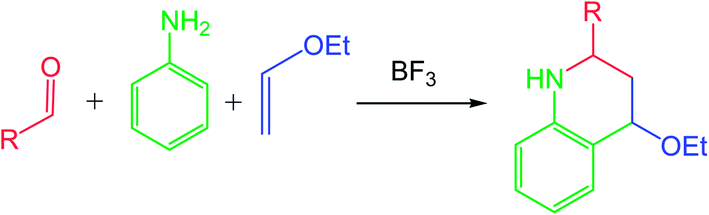 | ||
| Scheme 2 One-pot three-component Povarov reaction. | ||
Over the past decade, various advances in the study of PR have been reported,4 including the broadening of its scope (with respect to its components and the nature of the catalyst), progressing toward both the understanding of its reaction mechanism and its application in total synthesis.5 Due to the versatility in the functionalisation of the carbonyl derivative and the aniline, even the use of poor nucleophilic alkenes such as isoprene, together with the possibility of performing the PR in an intramolecular manner,6 have allowed this reaction to become a powerful tool for the construction of the quinoline structure.
1- and 2-azabutadienes display poor reactivity in polar Diels–Alder (P-DA) reactions. They must be electrophilically activated by the coordination with a LA to participate in the P-DA reaction toward nucleophilic alkenes.7 In addition, N-aryl imines such as 1 have a poor reactive aromatic ring participating along the cycloaddition, a behaviour that could reverse the addition reaction. These two behaviours demand a strong electrophilic activation of the N-aryl imine and the use of nucleophilic ethylenes in order to perform the reaction under mild conditions. For the electrophilic activation, quantitative amounts of strong LAs, and even Brønsted acids8 able to protonate the imine nitrogen, are used.
PR has a very complex mechanism and several approaches can be investigated. In a first approach, the PR can be considered as a domino reaction that is initialised by a LA catalysed A-DA reaction between the N-aryl imine 5 and a nucleophilic ethylene 6 to yield the formal [4 + 2] cycloadduct 7, which undergoes a 1,3-hydrogen shift to yield the final tetrahydroquinoline 8 (see Scheme 3).
 | ||
| Scheme 3 The simplified proposed mechanism for the Povarov reaction. | ||
However, the actual mechanism can be more complex and it can depend on the structural features of the N-aryl imine, the ethylene system or even the LA catalyst. Firstly, the use of strong electrophilically activated azadienes and strong nucleophilically activated ethylenes can turn an one-step A-DA reaction into a two-step one, with the participation of zwitterionic intermediates.9 Secondly, several theoretical studies have emphasised that tautomerisation via a 1,3-hydrogen shift is energetically very unfavourable due to the strain associated with the formation of a four-membered transition state (TS).10 Consequently, tautomerisation must be also stepwise with the participation of an acid/base species.
In this line, several experimental works have been devoted to establish the stepwise nature of the reaction mechanism, through the characterisation of feasible intermediates.11,12 For instance, Whiting proposed that formation of the product 11 from the reaction of N-aryl-C-ethoxycarbonyl imine 9 with the ER butadiene 10 could proceed from the hydrolysis of the zwitterionic intermediate 12 which would come from the nucleophilic attack of the non-substituted carbon of the ER butadiene 10 on the corresponding LA:imine complex of 9.12 Note that this nucleophilic addition corresponds to the first step of a stepwise P-DA reaction (see Scheme 4).
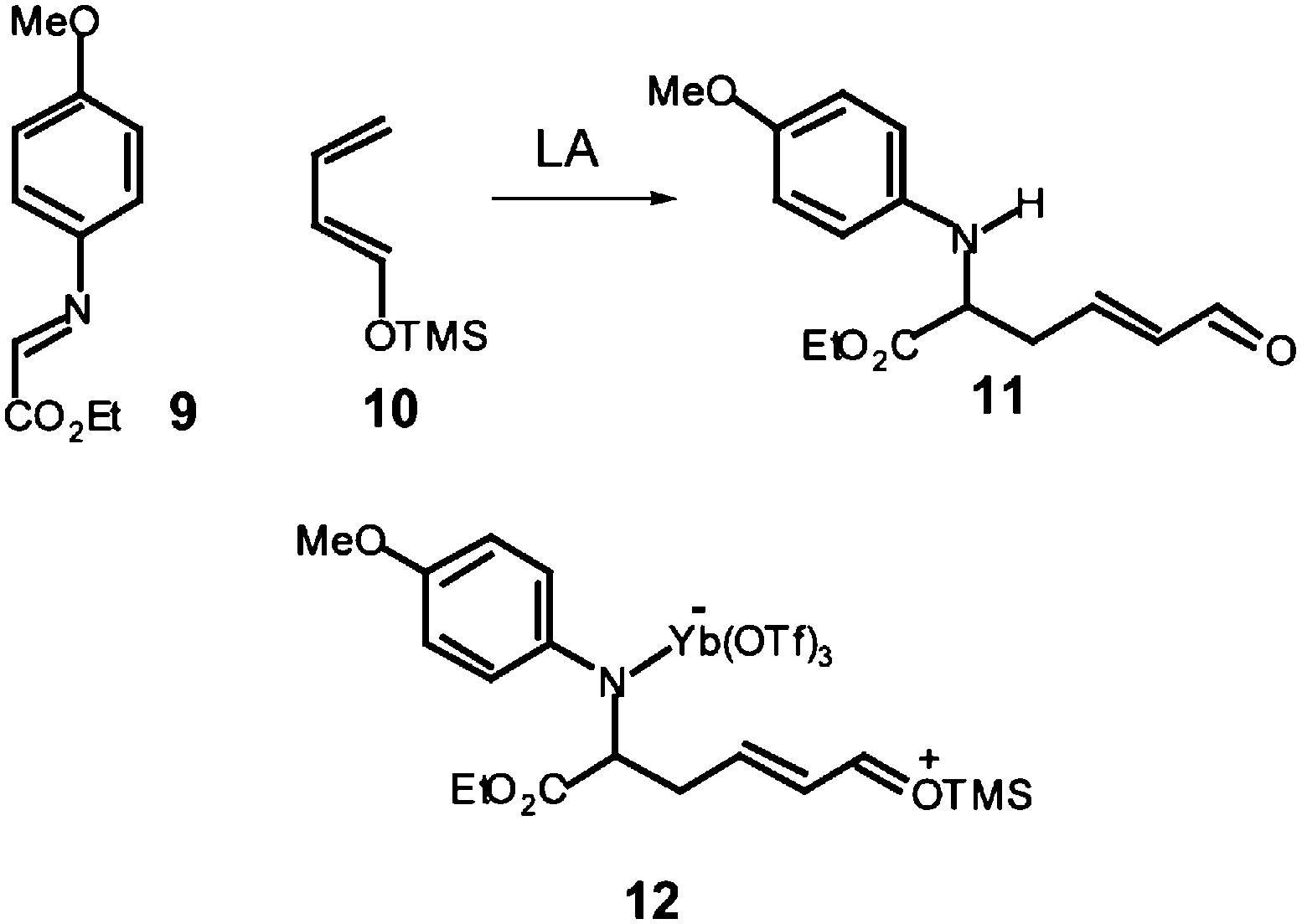 | ||
| Scheme 4 | ||
Very few theoretical studies devoted to the PR have been reported in the literature. In 2010, Cossío et al. studied the BF3 catalysed A-DA reactions of N-(3-pyridyl) imines such as 13b and 13c with ethylene, 14a, and substituted ethylenes such as styrene 14b as the first reaction of the domino Povarov sequence (see Scheme 5).13 At the B3LYP/6-31G* level, these A-DA reactions presented in gas phase one-step mechanisms through asynchronous TSs.13
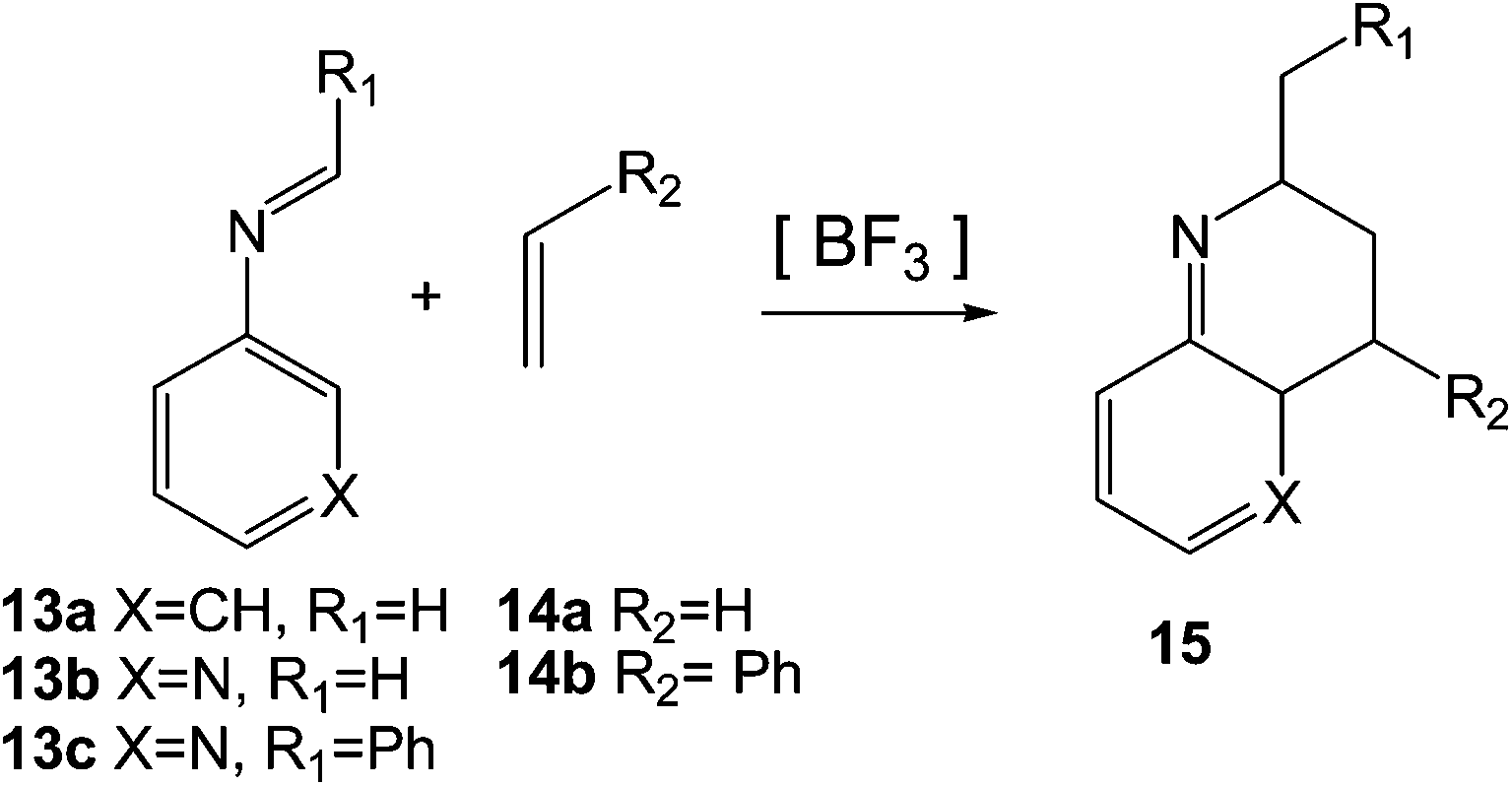 | ||
| Scheme 5 A-DA reactions of N-aryl imines 13a–c with ethylene 14a and styrene 14b. | ||
For the A-DA reactions of N-aryl imines 13a–c with ethylene 14a very high activation energies were found, between 27.0 and 32.9 kcal mol−1. The authors suggested that these disfavoured reactions may be a consequence of the significant decrease in the aromatic stabilisation energy in the TS.13 However, the low global electrophilicity ω index14 of N-aryl imine 13a and N-(3-pyridyl) imine 13b, ω = 1.24 and 1.63 eV, respectively, and the low global nucleophilicity N index15 of ethylene 14a, N = 1.86 eV, are responsible for the high activation energies involved in these non-polar Diels–Alder (N-DA) reactions16 (see below). The coordination of BF3 to N-aryl imines such as 13a and N-(3-pyridyl) imines such as 13b decreased the activation energies of the A-DA reactions to 18.8–30.9 kcal mol−1 as a consequence of the high electrophilic character of the corresponding BF3:imine complexes (13b:BF3 ω = 2.56 eV), but after all, they remained very high due to the low nucleophilic character of ethylene 14a. Note that the PR requires the use of ER ethylenes such as ethyl vinyl ether 2 (see Scheme 1).
The BF3 catalysed A-DA reactions of N-aryl and N-(3-pyridyl) imines 13a–c with substituted ethylenes such as styrene 14b presented lower activation energies as a consequence of the larger nucleophilic character of these substituted ethylenes when compared with ethylene 14a. Note that while ethylene 14a is classified as a poor nucleophile within the nucleophilicity scale,17 styrene 14b (N = 3.09 eV) is classified as strong nucleophile. Coordination of two BF3 molecules to N-(3-pyridyl) imines 13b–c decreased the activation energies as a consequence of the strong increase of the electrophilic character of the corresponding LA complex (ω = 3.30 eV). All these B3LYP/6-31G* LA catalysed A-DA reactions also presented one-step mechanisms in gas phase through highly asynchronous TSs.13
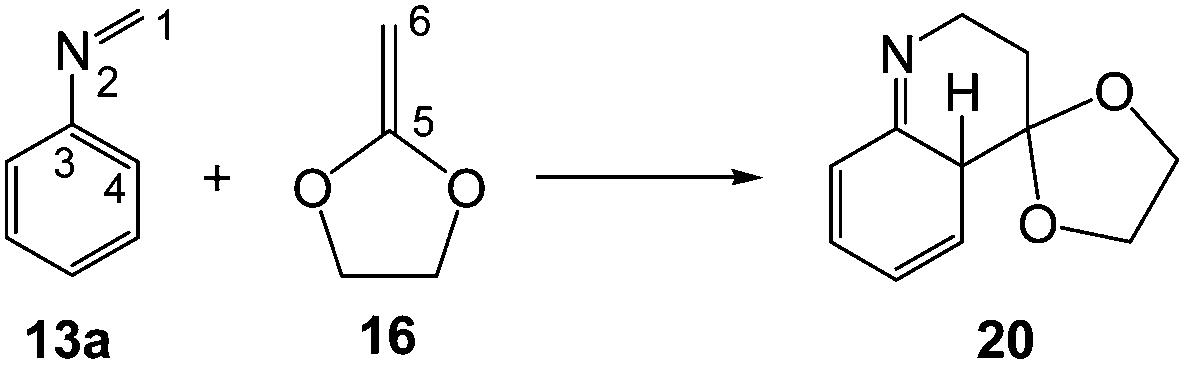
In the present work, we perform a complete characterisation of the reaction mechanism of the PR, as well as the main factors controlling the activation energy associated with the LA catalysed A-DA reaction involved in these domino reactions. For this purpose, a complete characterisation of the molecular mechanism of the PR between the simplest N-aryl imine 13a and 2-methylene-1,3-dioxolane 16, a strong nucleophilic ethylene, to yield tetrahydroquinoline 17 was performed (see Scheme 6). The LA catalysed A-DA reactions of N-aryl-C-aryl imine 1 and N-phenyl-C-ethoxycarbonyl imine 26 with methyl vinyl ether (MVE) 23, were also studied as models for experimental reactions, and the results were compared to those of the reaction model given in Scheme 6. Finally, an electron localisation function18 (ELF) topological analysis of some selected points along the intrinsic reaction coordinate19 (IRC) curve of the 13a + 16 reaction was carried out in order to characterise the bonding changes along this LA catalysed A-DA reaction.
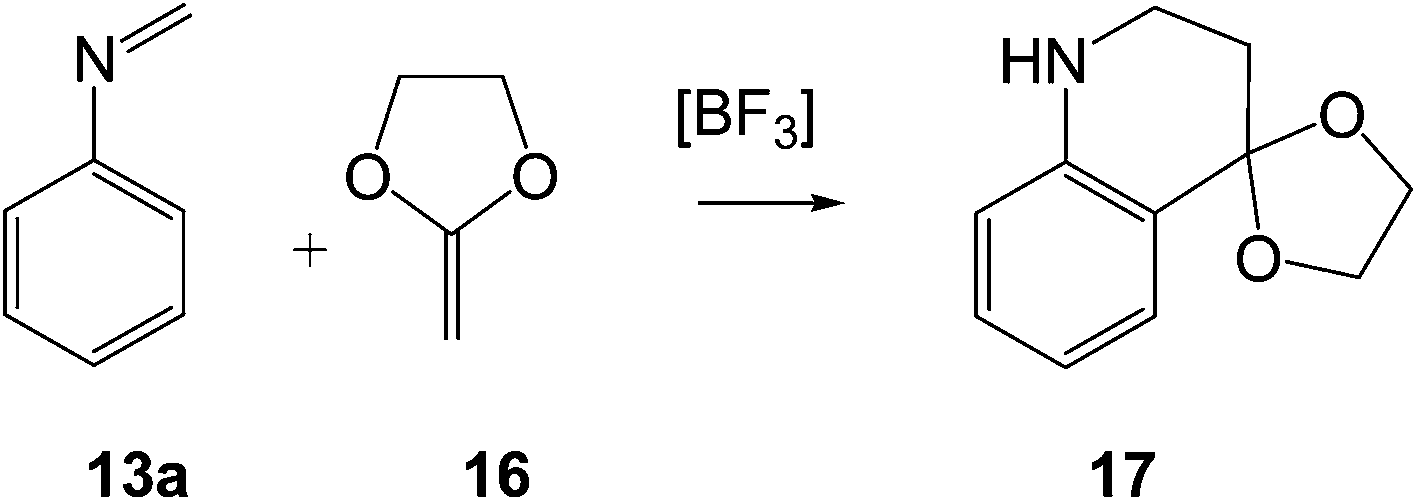 | ||
| Scheme 6 Reaction model for the Povarov reaction. | ||
Computational methods
Several works have shown that the B3LYP functional is relatively accurate for kinetic data, although the reaction exothermicities are underestimated.20 Recently, the Truhlar's group has proposed some functionals, such as the MPWB1K,21 which improve thermodynamic calculations. Due to that PRs are domino processes involving the formation of a cycloadduct intermediate, accurate thermodynamic calculations are required in order to understand the overall energy profile. Consequently, DFT computations were carried out using the MPWB1K21 exchange-correlation functional, together with the standard 6-311G** basis set.22 The optimisations were carried out using the Berny analytical gradient optimisation method.23 The stationary points were characterised by frequency computations in order to verify that TSs have one and only one imaginary frequency. The IRC paths19 were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González-Schlegel integration method.24 Solvent effects of acetonitrile in the optimisations were taken into account using the polarisable continuum model (PCM) as developed by Tomasi's group25 in the framework of the self-consistent reaction field (SCRF).26 Values of enthalpies, entropies and free energies in acetonitrile were calculated with the standard statistical thermodynamics at 25.0 °C and 1 atm.22 Thermodynamic calculations were scaled by a factor 0.96.27 The electronic structures of stationary points were analysed by the natural bond orbital (NBO) method28 and by the ELF topological analysis, η(r).18 The ELF study was performed with the TopMod program29 using the corresponding monodeterminantal wavefunctions of the selected structures of the IRC. All computations were carried out with the Gaussian 09 suite of programs.30The global electrophilicity index,14 ω, is given by the following expression, ω = (μ2/2η), in terms of the electronic chemical potential μ and the chemical hardness η. Both quantities may be approached in terms of the one-electron energies of the frontier molecular orbital HOMO and LUMO, εH and εL, as μ ≈ (εH + εL)/2 and η ≈ (εL − εH), respectively.31 Recently, we introduced an empirical (relative) nucleophilicity index,15 N, based on the HOMO energies obtained within the Kohn–Sham scheme,32 and defined as N = EHOMO(Nu)−EHOMO(TCE). The nucleophilicity is referred to tetracyanoethylene (TCE), because it presents the lowest HOMO energy in a large series of molecules already investigated in the context of polar cycloadditions. This choice allows us to handle conveniently a nucleophilicity scale of positive values. The P+k electrophilic and P−k nucleophilic Parr functions,33 which allow for the characterisation of the electrophilic and nucleophilic centers of a molecule, were obtained through the analysis of the Mulliken atomic spin density of the radical anion and the radical cation of the studied molecules, respectively.
Results and discussion
The present theoretical study has been divided into five parts: (i) firstly, a complete characterisation of the molecular mechanism of the PR between the simplest N-aryl imine 13a and 2-methylene-1,3-dioxolane 16 is performed; (ii) secondly, the LA catalysed A-DA reaction of N-aryl-C-aryl imine 1 with MVE 23, is studied as the prototype of the experimental PR; (iii) in the third part, the role of the substituent present in the imine C1 carbon is analysed; (iv) in the fourth part, an ELF topological analysis of selected points along the A-DA reaction of the simplest N-aryl imine 13a with the nucleophilically activated ethylene 16 is carried out in order to characterise the bonding changes along these LA catalysed A-DA reactions; and (v) finally, an analysis of the DFT-based reactivity indices of the reagents involved in PRs is performed in order to explain both the reactivity in these A-DA reactions and the complete regioselectivity experimentally observed.(i) Study of the mechanism of the PR between the simplest N-aryl imine 13a and 2-methylene-1,3-dioxolane 16
In order to perform a complete characterisation of the molecular mechanism of the LA catalysed PR, the BF3 catalysed domino reaction between the simplest N-aryl imine 13a and 2-methylene-1,3-dioxolane 16 was firstly studied. Note that 2-methylene-1,3-dithiolanes have been used experimentally as ER ethylenes in BF3 catalysed A-DA reactions.34 The BF3 promoted domino reaction between N-aryl imine 13a and the ER ethylene 16 takes place via a two-step A-DA reaction followed by a stepwise 1,3-hydrogen shift (see Scheme 7). The relative energies of the stationary points involved in this PR are depicted in Scheme 7. The total energies are given in Table S1 of the ESI.† A schematic representation of the energy profile of the PR between N-aryl imine 13a and 2-methylene-1,3-dioxolane 16 is given in Fig. 1.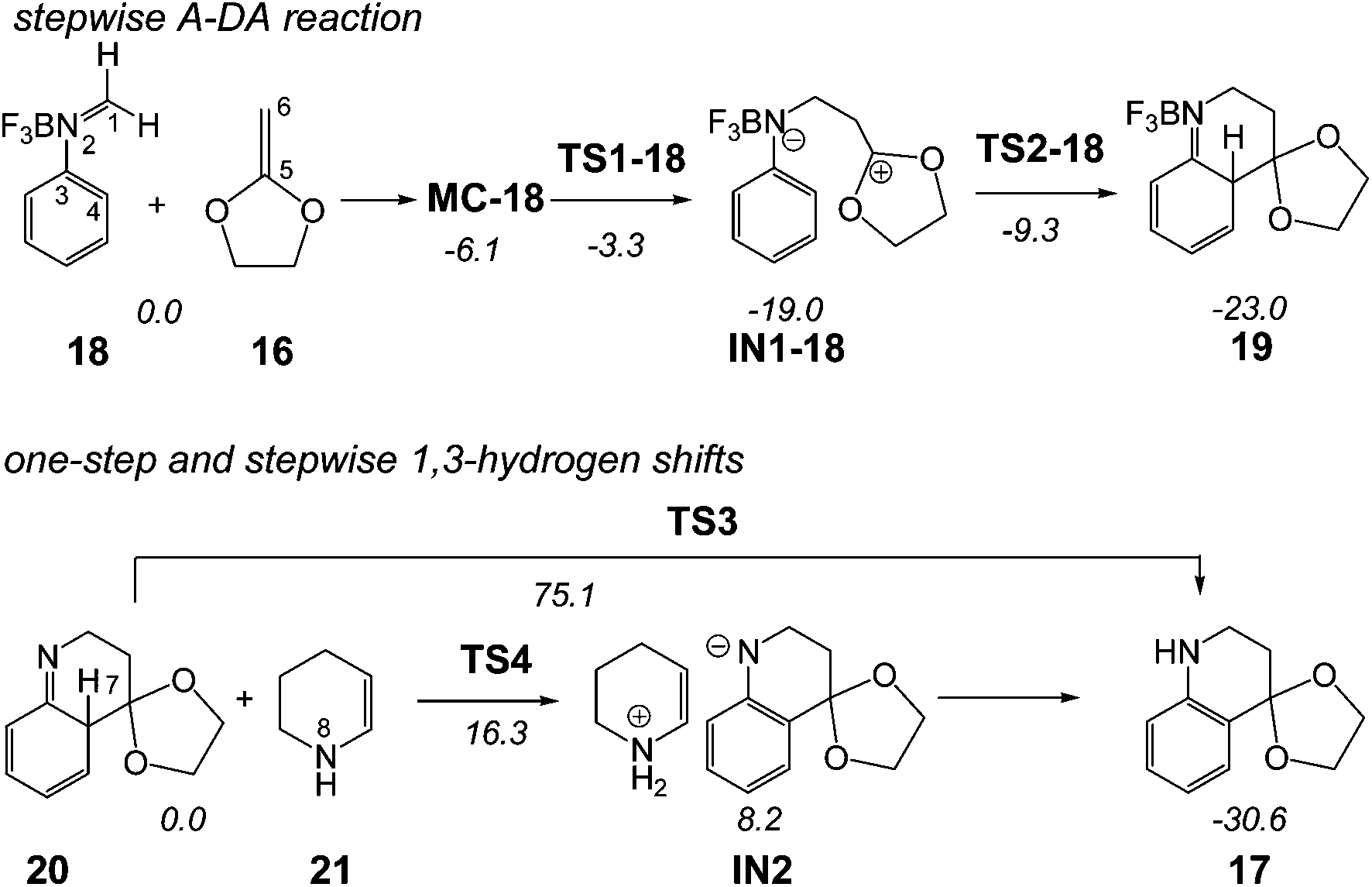 | ||
| Scheme 7 Povarov reaction between N-aryl imine 13a and 2-methylene-1,3-dioxolane 16. The MPWB1K/6-311G** relative energies (in kcal mol−1) in acetonitrile are in italics. | ||
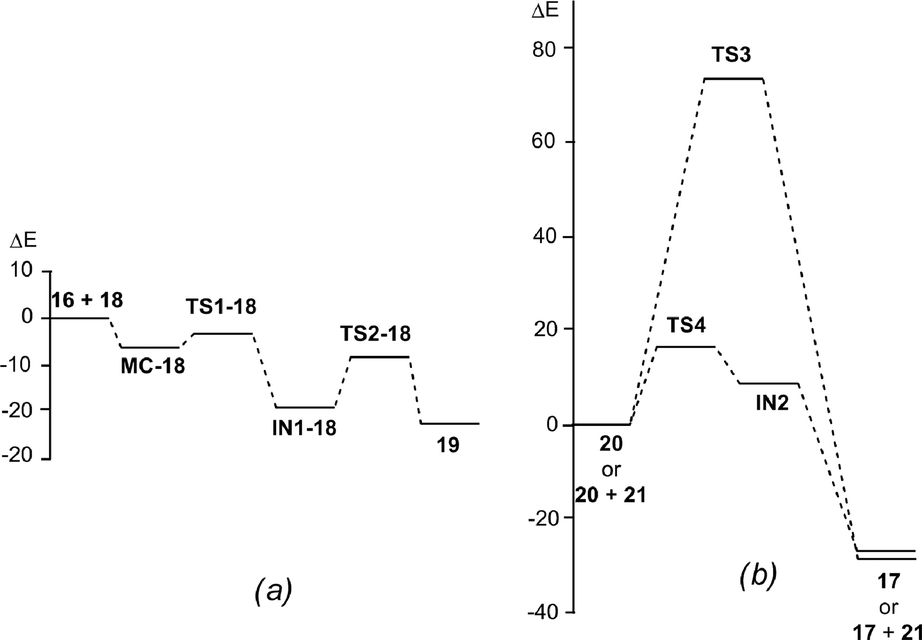 | ||
| Fig. 1 Energy profile (ΔE in kcal mol−1) of the Povarov reaction between N-aryl imine 13a and 2-methylene-1,3-dioxolane 16: (a) stepwise A-DA reaction; (b) one-step and stepwise 1,3-hydrogen shifts. | ||
The A-DA reaction is initialised by the nucleophilic attack of the ER ethylene 16 on the BF3:imine complex 18. For this nucleophilic attack, two regioisomeric channels are feasible. However, an analysis of the Parr functions33 allows ruling out the reaction path associated with the nucleophilic attack of the C6 carbon of ER ethylene 16 on the C4 carbon of 18 (see below). The TS associated with the nucleophilic attack of the C6 carbon of ethylene 16 on the C1 carbon of 18, TS1-18, is located 3.3 kcal mol−1 below the separated reagents. However, if we consider the formation of a molecular complex, MC-18, in an earlier step of the reaction, the activation energy becomes positive, 2.8 kcal mol−1. Interestingly, formation of the zwitterionic intermediate IN1-18 is strongly exothermic, −19.0 kcal mol−1. The second step of the A-DA reaction is the formation of the second C4–C5 single bond by a ring-closure process. The activation energy associated with this process via TS2-18 is 9.7 kcal mol−1. This barrier is higher than that associated with two-step P-DA reactions, as a consequence of the loss of the aromatic character of the phenyl ring. However, in spite of this fact, formation of compound 19 is strongly exothermic, −23.0 kcal mol−1.
Inclusion of thermal corrections and entropies to electronic energies rise the relative free energies between 13–18 kcal mol−1, due to the unfavourable entropy associated with the bimolecular reaction (see Table S5 in ESI†). Thus, formation of MC-18 is endergonic by 7.3 kcal mol−1. Consequently, MC-18 does not have any significant impact on the free energy profile. The free activation energy associated with TS1-18 is 12.8 kcal mol−1, and formation of IN1-18 is exergonic by 0.8 kcal mol−1. Finally, formation of compound 19 is exergonic by 5.7 kcal mol−1.
The second reaction of this domino process is a formal 1,3-hydrogen shift, allowing the conversion of compound 20 into the thermodynamically more stable tautomer 17, in which the aromatic ring is regenerated. Several theoretical studies have shown that the direct one-step 1,3-hydrogen shift for tautomerisations is very unfavourable due to the formation of a very strained four membered TS.35 Accordingly, the TS associated with the intramolecular tautomerisation of compound 20 into the final product 17 via TS3, presents a very high energy, 75.1 kcal mol−1, ruling out the direct 1,3-hydrogen shift. Formation of the final tetrahydroquinoline 17 from 20 is strongly exothermic, −30.6 kcal mol−1; tautomerisation being thermodynamically very favourable.
Due to the acidic character of the hydrogen involved in the tautomerisation, this process can be acid/base catalysed by any basic species present in the reaction.10 Recently, the effects of polar protic solvents such as water and methanol in the tautomerisation of pyridazin-3(2H)-one have been studied.36 The activation energy associated with the four membered TS in water decreased from 44.4 kcal mol−1 to 15.0 kcal mol−1 in the presence of an implicit water molecule. A more basic molecule such as tetrahydroquinoline 20 could decrease this activation energy, but now the tauromerization becomes stepwise. To model the intermolecular process, we selected tetrahydropyridine 21, which would model the basic character of tetrahydroquinoline 20. Now, the TS associated with the proton abstraction from 20 through the basic enamine 21, via TS4, was found 16.3 kcal mol−1 above the hydrogen-bonded complex 20 + 21, the formation of the zwitterionic intermediate IN2 being endothermic by 8.2 kcal mol−1. The subsequent step of this stepwise process is the cession of the proton present in the ammonium cation of 21 to the N2 nitrogen atom of 20. This acid/base process did not present any appreciable barrier. Therefore, when the two ionic species are approached, the proton transfer process to yield 17 + 21 would take place easily and quickly. Inclusion of thermal corrections and entropies to electronic energies rise the relative free energy of TS4 to 15.2 kcal mol−1, the tautomerization being exergonic by 31.9 kcal mol−1 (see Table S5 in ESI†). These data indicate that this tautomerisation is kinetically and thermodynamically very favourable.
The geometries of the TSs associated with the BF3 catalysed PR between N-aryl imine 13a and the ER ethylene 16 are given in Fig. 2. At TS1-18 the distance between the C1 and C6 carbons is 2.240 Å, while the distance between the C4 and C5 carbons is 3.224 Å. At intermediate IN1-18, the length of the C1–C6 single bond is 1.560 Å, while the distance between the C4 and C5 carbon remains at 3.117 Å. Interestingly, the C1–C6 length in intermediate IN1-18, which is shorter than that in most of the zwitterionic intermediates involved in two-step P-DA reactions (mainly between 1.60–1.65 Å),37 is in complete agreement with the high stabilisation of this intermediate. At TS2-18, associated with the ring-closure, the distance between the C4 and C5 carbons is 2.129 Å.
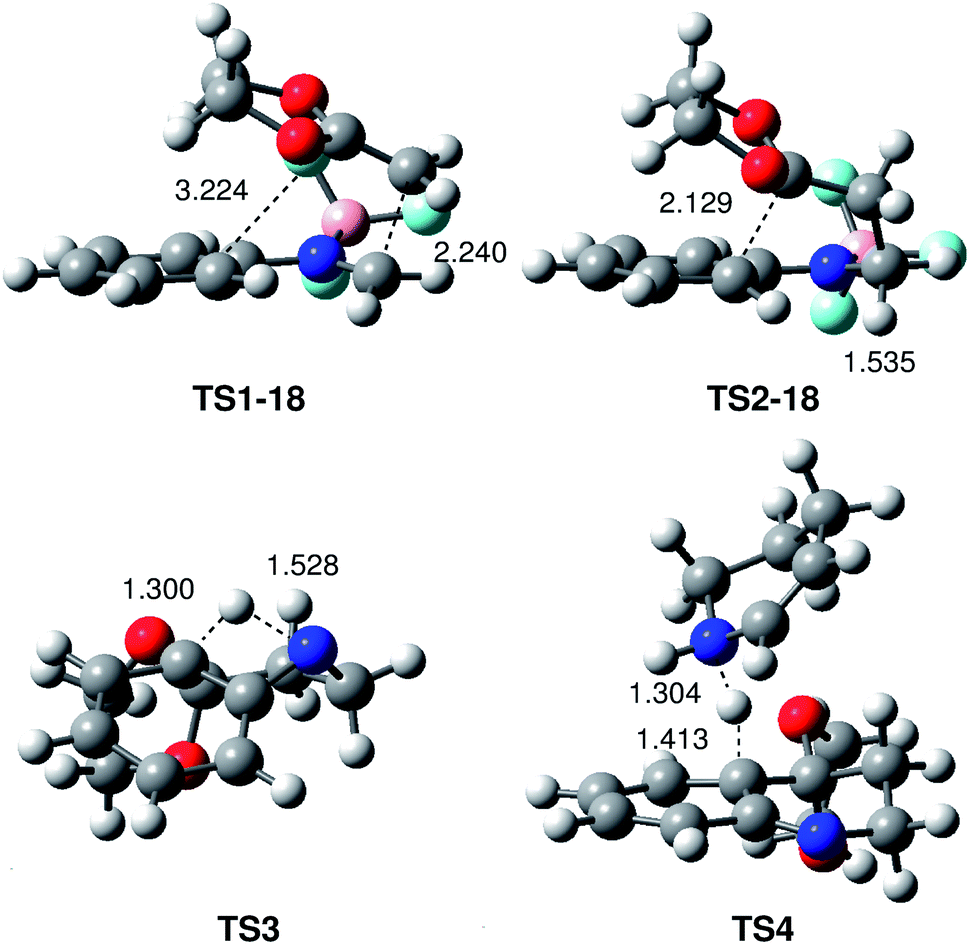 | ||
| Fig. 2 MPWB1K/6-311G** geometries, in acetonitrile of the transition structures involved in the Povarov reaction between the simplest N-aryl imine 13a and 2-methylene-1,3-dioxolane 16. Lengths are given in Angstroms. | ||
At TS3, associated with the intramolecular 1,3-hydrogen shift, the lengths of the C4–H7 breaking and N2–H7 forming bonds are 1.300 and 1.528 Å, respectively. At TS4, associated with the first step of the intermolecular 1,3-hydrogen shift, the length of the C4–H7 breaking bond is 1.413 Å, while the length of the H7–N8 forming bond is 1.304 Å (see Scheme 7 for atom numbering).
The polar nature of the BF3 catalysed A-DA reaction was analysed by computing the global charger transfer (GCT) along the cycloaddition. The natural atomic charges at TS1-18, IN1-18 and TS2-18, obtained through a natural population analysis (NPA),38 were shared between the ER ethylene and the LA:imine complex frameworks. The GCT that fluxes from the ER ethylene framework to LA:imine complex framework along this A-DA reaction is 0.29e at TS1-18, 0.86e at IN1-18 and 0.56e at TS2-18. These values confirm the high polar character of this A-DA reaction. NPA analysis at IN1-18 indicates the high zwitterionic character of this intermediate. At TS2-18, there is a decrease of the GCT as consequence of a retro-donation along the ring-closure process.
Finally, the A-DA reaction between the simplest N-aryl imine 13a and the ER ethylene 16 was studied in order to establish the role of the LA catalyst in the PR (see Scheme 8). Energetic and geometrical details of the mechanistic study are given in the ESI.†
 | ||
| Scheme 8 A-DA reaction between N-aryl imine 13a and ER ethylene 16. | ||
Interestingly, this uncatalysed reaction also presents a two-step mechanism. The activation energy associated with the nucleophilic attack of the ER ethylene 16 on N-aryl imine 13a, is 12.0 kcal mol−1, while the formation of the corresponding zwitterionic intermediate is found endothermic by 9.4 kcal mol−1. Consequently, the presence of the LA in the PR, does not only accelerate the reaction, but also permits the A-DA reaction to take place via a strong stabilised zwitterionic intermediate. Note that the formation of the intermediate IN1-18 is strongly exothermic, –19.0 kcal mol−1. This behaviour allows explaining that under adequate experimental conditions the corresponding intermediate could be isolated (see below).12
(ii) Study of the mechanism of the BF3 catalysed A-DA reactions of N-aryl-C-aryl imine 1 with methyl vinyl ether 23
Given that the formal 1,3-hydrogen shift is expected to be similar across PRs, only the BF3 catalysed A-DA reaction between the experimental N-aryl-C-aryl imine 1 and MVE 23 was studied as a reaction model of the experimental PR shown in Scheme 1. Due to the asymmetry of both reactants, this A-DA reaction can take place along four competitive channels: two stereoisomeric channels (the endo and exo ones) and two regioisomeric channels. Just as the A-DA reaction between the simplest N-aryl imine 13a and the ER ethylene 16, the study of the regioisomeric channels associated with the nucleophilic attack of the C6 carbon of MVE 23 on the C4 carbon of N-aryl-C-aryl imine 1 were ruled out as these channels were considered very unfavourable.9 Consequently, only the endo and exo stereoisomeric channels associated with the initial formation of the C1–C6 single bond were considered. Analysis of the stationary points associated with the formation of the formal [4 + 2] cycloadducts 24 and 25 indicates that this A-DA reaction also takes place along a two-step mechanism. Therefore, two TSs, a zwitterionic intermediate and the formal [4 + 2] cycloadduct were located and characterised along each one of the two stereoisomeric channels (see Scheme 9). The relative energies of the stationary points involved in the BF3 catalysed A-DA reaction between N-aryl-C-aryl imine 1 and MVE 23 are collected in Scheme 9. The total energies are given in Table S2 of the ESI.† A schematic representation of the energy profile of the BF3 catalysed A-DA reactions of N-aryl-C-aryl imine 1 with MVE 23 is given in Fig. 3.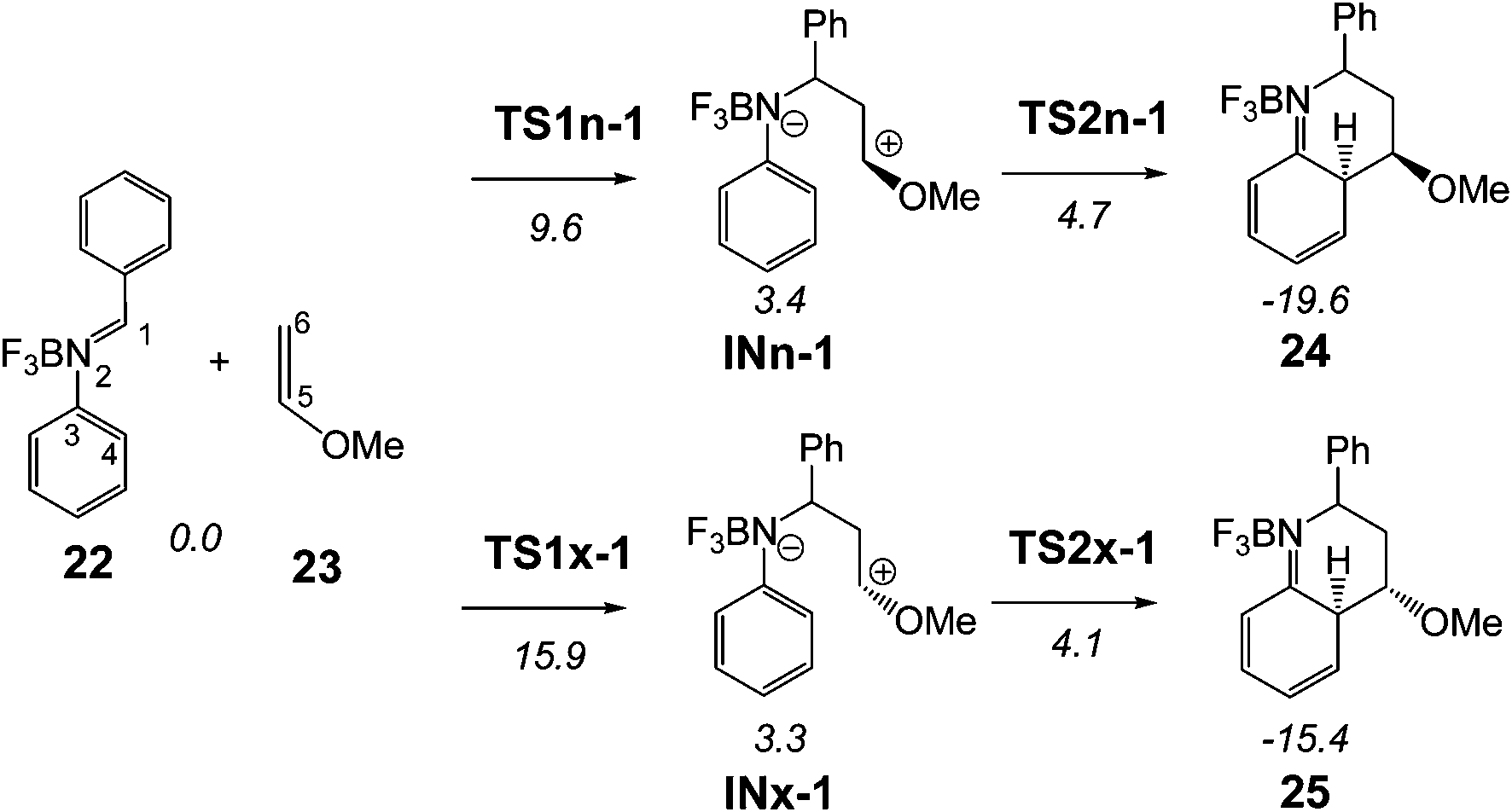 | ||
| Scheme 9 BF3 catalysed A-DA reaction between N-aryl-C-aryl imine 1 and MVE 23. The MPWB1K/6-311G** relative energies (in kcal mol−1) in acetonitrile are in italics. | ||
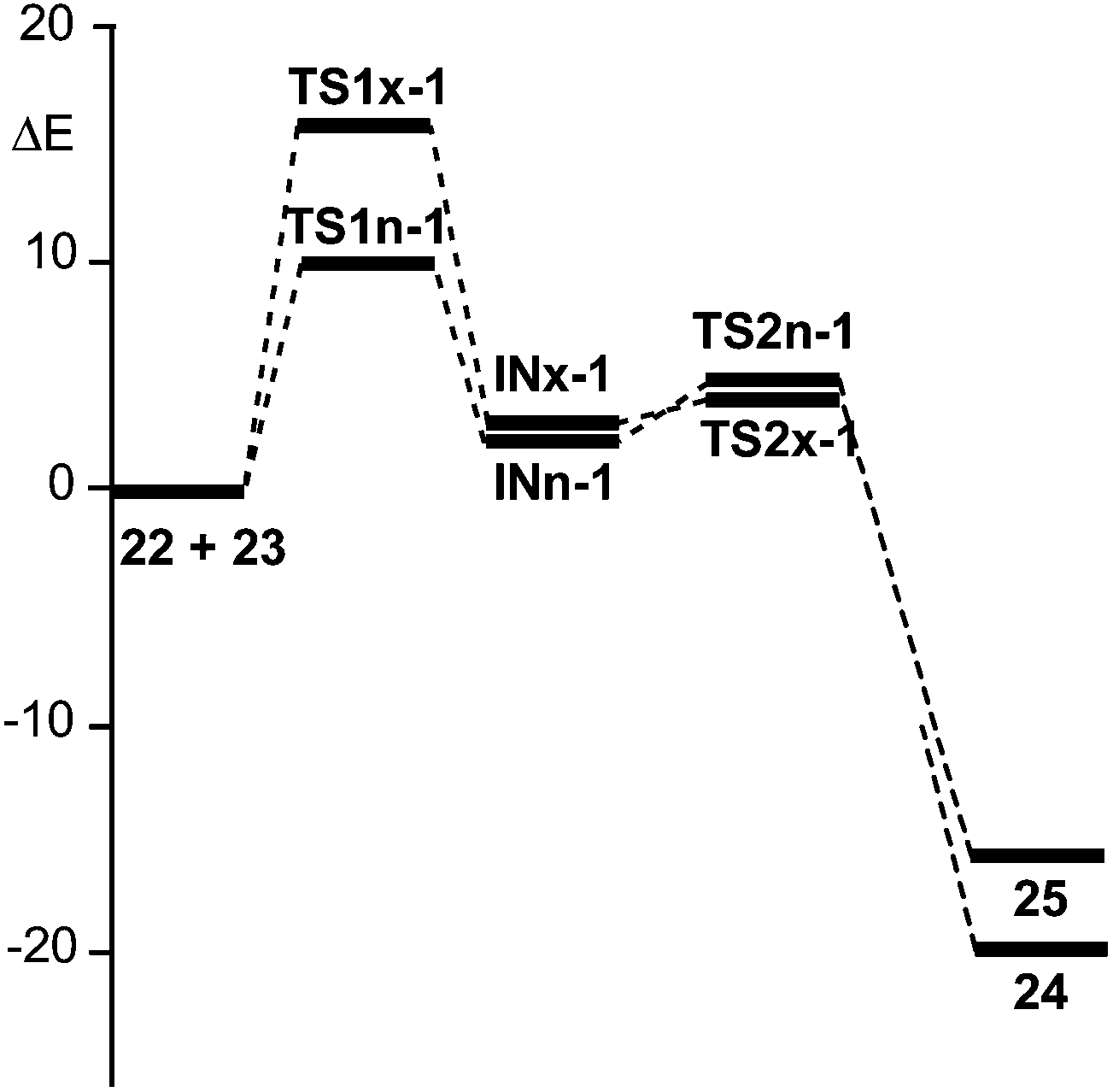 | ||
| Fig. 3 Energy profile (ΔE in kcal mol−1) of the BF3 catalysed A-DA reactions of N-aryl-C-aryl imine 1 with MVE 23. | ||
The BF3 catalysed A-DA reaction, between N-aryl-C-aryl imine 1 and MVE 23 to yield the formal [4 + 2] cycloadducts 24 and 25 begins with the nucleophilic attack of the C6 carbon of MVE 23 on the C1 carbon of BF3:imine complex 22. The activation energies associated with the two stereoisomeric nucleophilic attacks are 9.6 (TS1n-1) and 15.9 (TS1x-1) kcal mol−1. Formation of the corresponding zwitterionic intermediates is endothermic by 3.4 (INn-1) and 3.3 (INx-1) kcal mol−1. In this two-step A-DA reaction, formation of the second C–C single bond along the ring-closure process from the corresponding intermediate has a very low activation energy: 1.3 (TS2n-1) and 0.8 (TS2x-1) kcal mol−1. Formation of the formal [4 + 2] cycloadducts is exothermic by −19.6 (24) and −15.4 (25) kcal mol−1. Some appealing conclusions can be drawn from these energy results: (i) the activation energies associated with the formation of the first C1–C6 single bond are 12.9 (TS1n-1) and 19.2 (TS1x-1) kcal mol−1 higher than that for the A-DA reaction of the simplest N-aryl imine 13a with the ER ethylene 16, via TS1-18; (ii) this A-DA reaction is completely endo selective, TS1x-1 being 6.3 kcal mol−1 higher in energy than TS1n-1; (iii) while formation of the zwitterionic intermediate IN-18 is exothermic by −19.0 kcal mol−1, formation of INn-1 and INx-1 is slightly endothermic.
Such as the A-DA reaction of BF3:imine complex 18 with ER ethylene 16, inclusion of thermal corrections and entropies to electronic energies rise the relative free energies of the A-DA reaction of BF3:imine complex 22 with ER ethylene 23 between 16–21 kcal mol−1, due to the unfavourable entropy associated with the bimolecular reaction (see Table S5 in ESI†). Thus, the free activation energy associated with TS1n-1 and TS1x-1 rises to 28.3 and 31.8 kcal mol−1, respectively. In spite of the reduction of the relative free energies between the two stereoisomeric TSs, the A-DA reaction remains endo selective. Formation of the formal endo [4 + 2] cycloadduct 24 is endergonic by 1.1 kcal mol−1. Anyway, the strong exothermic character of the tautomerisations makes the Povarov reaction irreversible.
The geometries of the TSs associated with the BF3 catalysed A-DA reaction between N-aryl-C-aryl imine 1 and MVE 23 are given in Fig. 4. At TS1n-1 and TS1x-1, the distance between the C1 and C6 carbons is 2.002 and 1.963 Å, while the distance between the C4 and C5 carbons is 3.093 and 3.166 Å, respectively. At intermediates IN1n-1 and IN1x-1, the length of the C1–C6 single bond is 1.597 and 1.561 Å while the distance between the C4 and the C5 carbon remains to 2.803 and 2.744 Å. At TS2n-1 and TS2x-1, associated with the ring-closure process, the distance between the C4 and C5 carbons is 2.290 and 2.262 Å, respectively.
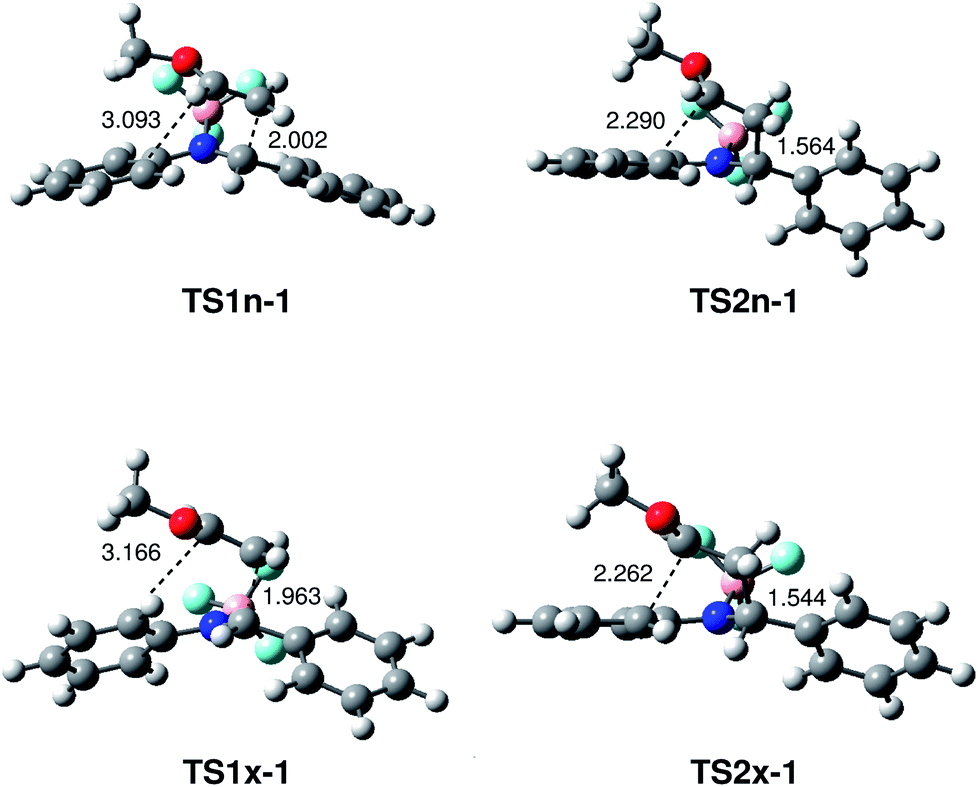 | ||
| Fig. 4 MPWB1K/6-311G** geometries of the transition structures involved in the BF3 catalysed A-DA reaction between N-aryl-C-aryl imine 1 and MVE 23. Lengths are given in Angstroms. | ||
The GCT that fluxes from the ER ethylene framework to BF3:imine complex one along the BF3 catalysed A-DA reaction of N-aryl-C-aryl imine 1 is 0.46e at both TS1n-1 and TS1x-1, 0.77e at IN1n-1, 0.81e at IN1x-1, 0.61e at TS2n-1 and 0.63e at TS2x-1. These values, which are similar to those found in the BF3 catalysed A-DA reaction of N-aryl imine 13a with the ER ethylene 16, confirm the high polar character of these BF3 catalysed A-DA reactions.
(iii) Study of the role of the C-substituent present in the imine, H, Ph, or CO2R, in the PR
Interestingly, the relative energy differences between TS1-18 and TS1n-1 with respect to the separated reagents, 12.9 kcal mol−1, is closer to that found between the TSs of the LA catalysed A-DA reactions between N-(3-pyridyl) imines 13b and 13c with styrene 14b, ca. 10 kcal mol−1 (see Scheme 5).13 Analysis of the structural behaviours of reagents participating in these A-DA reactions allows concluding that the main factor responsible for these energy differences could be related to the presence of the phenyl substituent in the C1 carbon of imines 1 and 13c, which is absent in the N-aryl imine 13a and 13b. Consequently, the loss of conjugation between the aryl substituent present in the C1 carbon of 1 and in 13c and the imine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group along the C1–C6 single bond formation, and not the decrease in the aromatic stabilisation energy in the TS such as proposed Cossío,13 is suggested to be responsible for these energy differences.
N group along the C1–C6 single bond formation, and not the decrease in the aromatic stabilisation energy in the TS such as proposed Cossío,13 is suggested to be responsible for these energy differences.
In order to establish the role of the substituent in the C1 carbon of imine 1, H, Ph or CO2R, two complementary studies were performed. First, the loss of conjugation was analysed using the isodesmic reactions39 given in Scheme 10 at the MPWB1K/6-311G** level.
 | ||
| Scheme 10 | ||
The energy difference associated with the isodesmic reaction involving the simplest N-aryl imine 13a, ΔEiso = −0.9 kcal mol−1, indicates that the conjugation of the aromatic N-aryl group with the imine CN double bond is thermodynamically slightly more unfavourable. However, an interesting result was found for N-aryl-C-aryl imine 1 (R = Ph) in which the energy difference associated with the corresponding isodesmic reaction, ΔEiso = 8.1 kcal mol−1, indicates that there is a substantial conjugation between the aromatic C-aryl substituent and the imine CN double bond. Note that for the N-aryl-C-aryl imine 1, the value of ΔEiso is close to the difference of activation energies between TS1-18 and TS1n-1, 12.9 kcal mol−1. Finally, the energy difference associated with the isodesmic reaction involving N-phenyl-C-ethoxycarbonyl imine 26 (R = CO2Me), ΔEiso = −0.6 kcal mol−1, is similar to that found for N-aryl imine 13a, indicating that the methoxycarbonyl substituent present at the C1 carbon atom of the imine CN group does not present any appreciable conjugation.
Alternatively, the endo reactive channel associated with the nucleophilic attack of MVE 23 on the BF3 complex of N-phenyl-C-ethoxycarbonyl imine 26, BF3:imine complex 27, was also studied in order to establish the role of the substituent present in the C1 atom of the imine (see Scheme 11). Energetic and geometrical details of the mechanistic study are given in the ESI.†
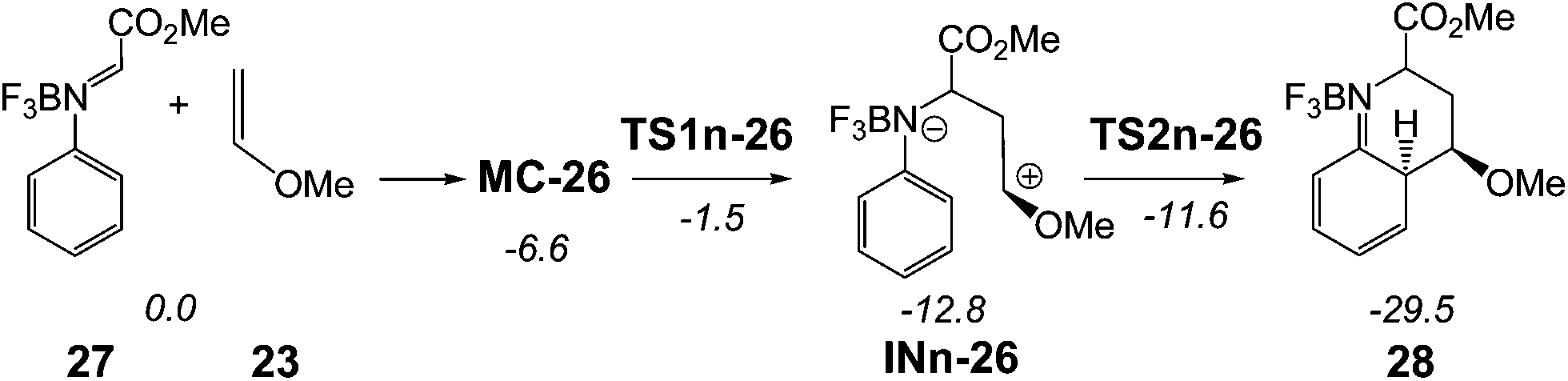 | ||
| Scheme 11 A-DA reaction between BF3:imine complex 27 and MVE 23. The MPWB1K/6-311G** relative energies (in kcal mol−1) in acetonitrile are in italics. | ||
Such as in the A-DA reaction shown in Schemes 7 and 9, the BF3 catalysed A-DA reactions of N-phenyl-C-ethoxycarbonyl imine 26 with MVE 23 presented also a two-step mechanism. In addition, this study allowed to find an anti zwitterionic intermediate, in agreement with the Whiting's proposal12 for the formation of the product 11 in Scheme 4 (see Scheme S2 in the ESI†).
Some appealing conclusions can be drawn from the energy results obtained from this study (see Table S4†): (i) when the phenyl group present in C1 is substituted by a methyl carboxylate group, the LA catalysed A-DA reaction presents a low activation energy, 5.1 kcal mol−1; (ii) in addition, this change in substitution makes the formation of the corresponding zwitterionic intermediates exothermic by more than −12 kcal mol−1; (iii) for the reaction involving N-phenyl-C-ethoxycarbonyl imine 26, the most favourable channel is that associated with the anti attack of MVE 23 on the imine carbon atom of 26; formation of the corresponding anti intermediate being kinetically and thermodynamically very favourable. These energy results provide an explanation for the formation of compound 11 observed by Whiting in the LA catalysed reaction between ethyl carboxylate derivative imine 9 and the ER diene 10 (see Scheme 4); and finally (iv) as the first step of these stepwise BF3 catalysed A-DA reaction is the rate-determining step of the cycloadditions, the presence of the N-aryl substituent does not have any incidence in the reaction rate since along the nucleophilic attacks of these ER ethylenes on the C1 carbon of the N-aryl BF3:imine complexes, the N-aryl substituent does not participate in the formation of the C1–C6 single bond. Consequently, this comparative analysis allows concluding that the C-substituent of the N-aryl imine has a more relevant role in the reaction rate that the N-aryl substituent due to the non participation of the N-aryl substituent in the rate determining step of these two-step BF3 catalysed A-DA reactions.
(iv) ELF topological analysis of bonding changes along the A-DA reaction between BF3:imine complex 18 and ER ethylene 16. A topological characterisation of the C–C bond formation in the PR
Several theoretical studies have shown that the ELF topological analysis along a reaction path can be used as a valuable tool to understand the bonding changes along the reaction path.40 In order to understand the C–C bond formation in the PR, an ELF topological analysis of the MPWB1K/6-31G* wave functions of some relevant points of the IRCs of the two-step A-DA reaction between BF3:imine complex 18 and ER ethylene 16 was carried out. Details of the ELF topological analysis are given in the ESI.†Some interesting conclusions can be drawn from the ELF topological analysis: (i) the C1–C6 single bond is formed at the distance of 1.884 Å (see P-I in Table S6†), by coupling of two pseudoradical centers41 appearing at the most nucleophilic and at the most electrophilic positions of the reagents (see later); (ii) formation of this pseudoradical centers are a consequence of the large GCT that takes place along this A-DA reactions. Note that at P-I the GCT is ca. 0.60e, (iii) at the zwitterionic intermediate IN1-18, while the C1–C6 single bond has already reached a 95% of the electron population present at the formal [4 + 2] cycloadduct 19, the formation of the second C4–C5 single bond has not started yet; (iv) formation of the second C4–C5 single bond takes place at the second step of the A-DA reaction at a C–C distance of 1.942 Å by coupling of two pseudoradical centers located at the C4 and C5 carbons; (v) this pattern for the formation of the two C–C single bonds along this stepwise A-DA reactions is similar to that observed in other P-DA reactions taking place along two-stage one-step mechanism.42 This behaviour permits establishing that both two-stage one-step and two-step mechanisms are non-concerted bond-formation processes.
This picture of the A-DA reaction given by the ELF topological analysis of bonding changes is opposed to that proposed by Cossío, who established that PRs with ER olefins take place in an asynchronous concerted process.13 It is interesting to note that most of the B3LYP/6-31G* TSs associated with the A-DA reaction of N-aryl imines 13 presented an asynchronous C–C bond formation in which the shorter C–C distance is below 1.90 Å, whereas the longer distance is greater than 2.40 Å. Considering that B3LYP/6-31G* ELF topological analyses of bonding changes along polar and non-polar DA reactions have established that the C–C single bond formation begins in the short range of 1.9–2.0 Å,42,43 these geometric parameters allow asserting that at these advanced asynchronous TSs, the formation of the first C–C single bond has already started; whereas the formation of the second C–C single bond has not started yet.
(v) Analysis of the global reactivity indices of the reagents involved in A-DA reactions
Finally, these A-DA reactions were analysed using the reactivity indices defined within the conceptual DFT.44 The values of global descriptors, namely, electronic chemical potential μ, chemical hardness η, global electrophilicity ω, and global nucleophilicity N indices for the series of reagents involved in these A-DA reactions are given in Table 1.| μ | η | ω | N | |
|---|---|---|---|---|
| 22 | −4.63 | 4.38 | 2.44 | 2.30 |
| 27 | −4.83 | 4.94 | 2.36 | 1.82 |
| 18 | −4.80 | 5.00 | 2.31 | 1.82 |
| 26 | −4.26 | 4.44 | 2.05 | 2.64 |
| 1 | −3.73 | 4.35 | 1.60 | 3.22 |
| 13a | −3.59 | 5.21 | 1.24 | 2.92 |
| 29 | −3.61 | 5.63 | 1.15 | 2.70 |
| 30 | −3.45 | 5.67 | 1.05 | 2.83 |
| 31 | −2.87 | 5.34 | 0.77 | 3.58 |
| 14a | −3.37 | 7.77 | 0.73 | 1.86 |
| 23 | −2.45 | 6.99 | 0.43 | 3.18 |
| 2 | −2.40 | 6.98 | 0.41 | 3.23 |
| 16 | −1.80 | 7.36 | 0.22 | 3.64 |
It turns out that the substitution of the C2 carbon of 1,3-butadiene 30 by a nitrogen atom does not modify substantially the reactivity indices of 2-azabutadiene 29 respect to those in 30. Thus, 2-azabutadiene 29 presents a low electrophilicity ω index, ω = 1.15 eV, being classified as a moderate electrophile,45 and a nucleophilicity N index of N = 2.70 eV, being also classified as a moderate nucleophile.17 N-aryl imine 13a is slightly more electrophilic, ω = 1.24 eV, and slightly more nucleophilic, N = 2.92 eV, than 2-azabutadiene 29. The inclusion of the second aryl group in the imine carbon provokes a larger effect; thus the experimental N-aryl-C-aryl imine 1 presents a higher electrophilicity index, ω = 1.60 eV, and a higher nucleophilicity index, N = 3.22 eV, being classified as a strong electrophile and a strong nucleophile. Finally, N-phenyl-C-ethoxycarbonyl imine 26 presents the largest electrophilic activation of the imine series, ω = 2.05 eV.
Coordination of the BF3 LA to the nitrogen atom of these imines provokes the most drastic increase of the electrophilicity ω index of the corresponding BF3:imine complexes between 0.31 and 1.07 eV. Indeed, the electrophilicity ω indices of these BF3 coordinated N-aryl imines are 2.31 eV (18), 2.36 eV (27) and 2.44 eV (22). This electrophilic activation accounts for the effects of the LA catalysts to allow the corresponding A-DA reaction to take place through a more polar process.16 It is interesting to note that in spite of the higher electrophilic character of N-aryl-C-aryl imine complex 22 than N-phenyl-C-ethoxycarbonyl imine complex 27, the former is less reactive.
In P-DA reactions, the GCT that takes place along the reaction is one of the main factors favouring the reaction, a behaviour that can be anticipated by analysing global and local reactivity indices of both reagents.16 However, other structural factors such the loss of conjugation along the reaction can disfavour a chemical process.46 Thus, in the case of N-aryl-C-aryl imine 22, the loss of conjugation between C-aryl and the imine frameworks along the reaction, which is not present in the other imines, appears to be one of the unfavourable factors. It is interesting to note that in spite of the fact that the BF3 complex of N-aryl-C-aryl imine 1 is more electrophilic than the BF3 complexes of N-phenyl-C-ethoxycarbonyl imine 26 and the simplest N-aryl imine 13a, the former is less reactive as a consequence of the presence of the C-aryl substituent. Note also that the polar character of the reaction, measured through the GCT at the TS associated with the nucleophilic attack of the ER ethylenes to the BF3:imine complexes, follows the trend of the electrophilicity index; 0.46e at TS1n-1, 0.33e at TS1n-26, and 0.29e at TS1-18.
P-DA reactions require the participation of good electrophiles and good nucleophiles. Ethylene 14a is one of poorest electrophile, ω = 0.73 eV, and nucleophile, N = 1.86 eV, species involved in DA reactions, being classified as marginal electrophile and marginal nucleophile. Consequently, it can not participate in P-DA reactions.43 Substitution of one hydrogen of ethylene by an electron-releasing methoxy group increases notably the nucleophilicity N index of MVE 23, N = 3.18 eV, and decreases slightly its electrophilicity ω index, ω = 0.43 eV. Consequently, MVE 23 is classified as a strong nucleophile participating in P-DA reactions.16 Note that MVE 23 has a similar nucleophilicity N index than the experimentally used ethyl vinyl ether 2, N = 3.23 eV. The inclusion of a second methoxy group in the ethylene framework increases notably the nucleophilicity N index of the ER ethylene 16, N = 3.64 eV, becoming the most nucleophilic species given in Table 1. Consequently, the A-DA reactions between the BF3:imine complexes with the ER ethylenes 16 and 23 will have a large polar character in complete agreement with the high GCT found at the corresponding TS.
Along a polar reaction involving the participation of asymmetric reagents, the most favourable reactive channel is that involving the initial two-center interaction between the most electrophilic center and the nucleophilic center of the two reagents. Recently, we have proposed the electrophilic P+k and nucleophilic P−k Parr functions derived from the excess of spin electron density reached via the GCT process from the nucleophile to the electrophile as powerful tools in the study of the local reactivity in polar processes.33 Accordingly, the electrophilic P+k Parr functions of BF3:imine complexes and nucleophilic P−k Parr functions of ER ethylenes 16 and 23 are analysed (see Fig. 5).
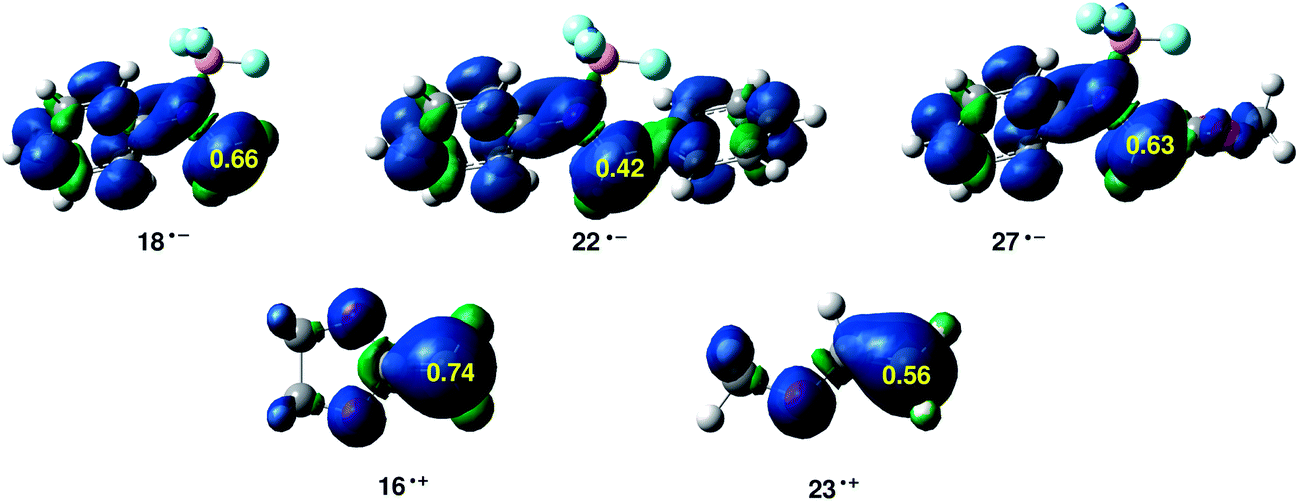 | ||
| Fig. 5 B3LYP/6-31G* Maps of the ASD (Atomic Spin Densities) of radical anions and electrophilic P+k Parr functions at the C1 carbon of BF3:imine complexes 18, 22 and 27, and maps of the ASD of the radical cations and nucleophilic P−k Parr functions at the C6 carbon of ER ethylenes 16 and 23. | ||
At the BF3:imine complexes 18, 22 and 27, the C1 atom presents the maximum value of the electrophilic P+k Parr functions, indicating that this site is the most electrophilic center of these molecules. At these carbons, the value of the local electrophilicity ωk index is ωC1 = 1.52 eV (18), ωC1 = 1.03 eV (22) and ωC1 = 1.49 eV (27). Interestingly, in spite of the fact that the BF3:imine complex 22 is the most electrophilic species of this series, the C1 carbon of 22 presents the lowest local electrophilicity ωk value. This behaviour together with the loss of conjugation along the reaction can explain the lower reactivity of BF3:imine complex 22 in comparison with BF3:imine complexes 18 and 27.
At the ER ethylenes 16 and 23, the C6 carbon presents the maximum value of the nucleophilic P−k Parr functions, being the most nucleophilic centers of these ER ethylenes. At these carbons, the value of the local nucleophilicity Nk index is NC6 = 2.70 eV (16) and NC6 = 1.78 eV (23). Note that the local nucleophilicity NC6 index in the ER ethylene 16 is twice than that in MVE 23.
Consequently, the most favourable electrophile–nucleophile interaction along these A-DA reactions will be that between the most electrophilic center of these BF3:imine complexes, the C1 carbon, and the most nucleophilic center of these ER ethylenes, the C6 carbon. In addition, the high values of the Parr functions at these centers account for the high asynchronicity found at the corresponding TSs.
It should be noted the values of the electrophilic P+k Parr functions at the imine and carboxyl carbons in the BF3:imine complex 27. Very recently, we defined the most electrophilic center of an organic molecule as the center with the highest spin density achieved through the GCT process, and not, as expected, at the center that presents the largest positive charge.47 In this way, for BF3:imine complex 27, the electrophilic P+k Parr function at the C1 carbon is 0.63, while at the carbonyl carbon atom is −0.02. On the other hand, the Mulliken net atomic charges are −0.09e at C1 and 0.56e at the carbonyl carbon (see Chart 1). Consequently, these values reinforce that the local electrophilicity is not charge-controlled.47
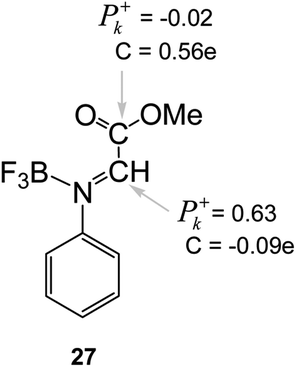 | ||
| Chart 1 Electrophilic P+k Parr functions and charges (C, in electrons) at the imine and carboxyl carbons of the BF3:imine complex 27. | ||
Conclusions
The molecular mechanism of the PR in acetonitrile solvent has been theoretically studied using the MPWB1K/6-311G** method. The PR is a domino process that comprises two sequential reactions: (i) a LA catalysed A-DA reaction between a N-aryl imine and an ER ethylene yielding a formal [4 + 2] cycloadduct; and (ii) a stepwise 1,3-hydrogen shift at this intermediate affording the final tetrahydroquinoline.The present study allows establishing a significant understanding about the PR mechanism. N-aryl imines do not experiment the PR due to the low electrophilic character of the CN imine group. Coordination of a LA to the imine nitrogen favours the PR towards ER ethylenes due to the increase of the electrophilicity of the corresponding BF3:imine complex that favours the A-DA reaction through a polar process. At the MPWB1K/6-311G** level, in acetonitrile, the LA catalysed A-DA reaction present a two-step mechanism as a consequence of the large stabilisation of the corresponding zwitterionic intermediate. The first step, which is associated with the nucleophilic attack of the ER ethylene on the imine carbon, constitutes the rate-determining step of these A-DA reactions. Interestingly, the presence of the aromatic N-aryl substituent in the imine has no remarkable incidence on the activation energy as it does not participate in the formation of the first C–C single bond. However, the present study establishes that the presence of a second C-aryl substituent has a relevant role in the activation energy as a consequence of the loss of conjugation along the nucleophilic attack on the imine carbon atom.
Although the formation of the second C–C single bond involves the aromatic ring of the N-aryl substituent, the subsequent formation of a very stable neutral species along the ring closure makes the corresponding A-DA reaction thermodynamically favourable. In addition, a further formal 1,3-hydrogen shift in the corresponding formal [4 + 2] cycloadduct, allowing the regeneration of the lost aromaticity in the A-DA step of the Povarov domino reaction permits the formation of the tetrahydroquinoline observed in the PR. Due to the high strain associated with the intramolecular 1,3-hydrogen shift, the tautomerisation must be acid/base catalysed.
An ELF topological analysis of bonding changes along the stepwise A-DA reaction between the BF3:imine complex 18 and the ER ethylene 16, indicates that the formation of the first C–C single bond takes place by C-to-C coupling of the two pseudoradical centers formed at the most nucleophilic center of the ER ethylene 16 and the most electrophilic center of complex 18, at a C–C distance of 1.88 Å. Formation of the second C–C single bond takes place at a C–C distance of 1.94 Å, after passing the second TS. This ELF topological analysis, which is similar to that found along the IRC of most P-DA reactions taking place along a two-stage one-step mechanism, permits establishing that P-DA reactions taking place along highly asynchronous TS are non-concerted processes.
Analysis of global and local DFT-based reactivity indices, performed at the ground state of the reagents, provides an additional explanation of the behaviours of the A-DA reaction involved in PRs. Analysis of the electrophilicity ω index of the N-aryl imines establishes the necessity to form a LA complex to increase the electrophilicity of the corresponding LA:imine complex in order to participate in a P-DA reaction. On the other hand, the use of nucleophilically activated ethylenes such as vinyl ethers is demanded to favour the amount of GCT required for these A-DA reactions.
Finally, analysis of the recently proposed electrophilic P+k and nucleophilic P−k Parr functions allows explaining the total regioselectivity found in PRs; while electrophilic P+k functions indicate that the imine carbon is the most electrophilic center of the BF3:imine complexes, the nucleophilic P−k Parr functions show that the non-substituted carbon is the most nucleophilic center of the ER ethylenes.
Acknowledgements
This work has been supported by the University of Valencia (project UV-INV-AE13-139082). J. A. S. also thanks CSIC contract JAE-Doc 2010-06204 co-funded by FSE.References
- L. S. Povarov, Russ. Chem. Rev., 1967, 36, 656 CrossRef PubMed.
- (a) Y. Ma, C. Qian, M. Xie and J. Sun, J. Org. Chem., 1999, 64, 6462 CrossRef CAS; (b) S. Kobayashi, T. Busujima and S. Nagayama, Synlett, 1999, 545 CrossRef CAS PubMed; (c) S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 120, 8287 CrossRef CAS; (d) S. Kobayashi, H. Ishitani and S. Nagayama, Synthesis, 1995, 1195 CrossRef CAS PubMed; (e) Y. Makioka, T. Shindo, Y. Taniguchi, K. Takaki and Y. Fuziwara, Synthesis, 1995, 801 CrossRef CAS PubMed; (f) S. Kobayashi, H. Ishitani and S. Nagayama, Chem. Lett., 1995, 423 CrossRef CAS; (g) S. Kobayashi, M. Araki, H. Ishitani, S. Nagayama and I. Hachiya, Synlett, 1995, 233 CrossRef CAS PubMed.
- (a) J. T. Shaw, Nat. Prod. Rep., 2009, 26, 11 RSC; (b) J. P. Nandy, M. Prakesch, S. Khadem, P. T. Reddy, U. Sharma and P. Arya, Chem. Rev., 2009, 109, 1999 CrossRef CAS PubMed; (c) S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS.
- For a review, see: V. A. Glushkov and A. G. Tolstikov, Russ. Chem. Rev., 2008, 77, 137 CrossRef CAS PubMed.
- (a) M. Hadden, M. Nieuwenhuyzen, D. Osborne, P. J. Stevenson, N. Thompson and A. D. Walker, Tetrahedron, 2006, 62, 3977 CrossRef CAS PubMed; (b) H. Twin and R. A. Batey, Org. Lett., 2004, 6, 4913 CrossRef CAS PubMed; (c) D. A. Powell and R. A. Batey, Org. Lett., 2002, 4, 2913 CrossRef CAS PubMed; (d) R. A. Batey and D. A. Powell, Chem. Commun., 2001, 2362 RSC.
- (a) B. V. Subba Reddy, A. Antony and J. S. Yadav, Tetrahedron Lett., 2010, 51, 3071 CrossRef PubMed; (b) A. A. Kudale, D. O. Miller, L. N. Dawea and G. J. Bodwell, Org. Biomol. Chem., 2011, 9, 7196 RSC.
- V. V. Kouznetsov, Tetrahedron, 2009, 65, 2721 CrossRef CAS PubMed.
- P. A. Grieco and A. Bahsas, Tetrahedron Lett., 1988, 29, 5855 CrossRef CAS.
- L. R. Domingo, Tetrahedron, 2002, 58, 3765 CrossRef CAS.
- R. Castillo, J. Andrés and L. R. Domingo, Eur. J. Org. Chem., 2005, 4705 CrossRef CAS.
- F. Linkert, S. Laschat, S. Kotila and T. Fox, Tetrahedron, 1996, 52, 955 CrossRef CAS.
- S. Hermitage, D. A. Jay and A. Whiting, Tetrahedron Lett., 2002, 43, 9633 CrossRef CAS.
- F. Palacios, C. Alonso, A. Arrieta, F. P. Cossío, J. Ezpeleta, M. Fuertes and G. Rubiales, Eur. J. Org. Chem., 2010, 2091 CrossRef CAS.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615 CrossRef CAS PubMed; (b) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168 RSC.
- L. R. Domingo and J. A. Sáez, Org. Biomol. Chem., 2009, 7, 3576 CAS.
- P. Jaramillo, L. R. Domingo, E. Chamorro and P. Pérez, J. Mol. Struct.: THEOCHEM, 2008, 865, 68 CrossRef CAS PubMed.
- (a) A. Savin, A. D. Becke, J. Flad, R. Nesper, H. Preuss and H. G. von Schnering, Angew. Chem., Int. Ed., 1991, 30, 409 CrossRef; (b) B. Silvi and A. Savin, Nature, 1994, 371, 683 CrossRef CAS; (c) A. Savin, B. Silvi and F. Colonna, Can. J. Chem., 1996, 74, 1088 CrossRef CAS; (d) A. Savin, R. Nesper, S. Wengert and T. F. Fassler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808 CrossRef CAS.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- (a) C. E. Check and T. M. Gilbert, J. Org. Chem., 2005, 70, 9828 CrossRef CAS PubMed; (b) G. O. Jones, V. A. Guner and K. N. Houk, J. Phys. Chem. A, 2006, 110, 1216 CrossRef CAS PubMed; (c) G. A. Griffith, I. H. Hillier, A. C. Moralee, J. M. Percy, R. Roig and M. K. Vicent, J. Am. Chem. Soc., 2006, 128, 13130 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- (a) H. B. Schlegel, J. Comput. Chem., 1982, 2, 214 CrossRef; (b) H. B. Schlegel, in Modern Electronic Structure Theory, ed. D. R. Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- (a) C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef; (b) C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853 CrossRef PubMed.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS; (b) B. Y. Simkin and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions-A Computational Approach, Ellis Horwood, London, 1995 Search PubMed.
- (a) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS PubMed; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- A. P. Sott and L. Radom, J. Phys. Chem., 1996, 100, 16502 CrossRef.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS PubMed; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS; (b) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev. [Sect.] A, 1965, 140, 1133 CrossRef.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486 RSC.
- D. Cheng, J. Zhou, E. Saiah and G. Beaton, Org. Lett., 2002, 4, 4411 CrossRef CAS PubMed.
- J. Andrés, L. R. Domingo, M. T. Picher and V. S. Safont, Int. J. Quantum Chem., 1998, 66, 9 CrossRef.
- S. R. Emamian, L. R. Domingo and S. F. Tayyari, J. Mol. Graphics Modell., 2014, 49, 47 CrossRef CAS PubMed.
- L. R. Domingo and J. Andrés, J. Org. Chem., 2003, 68, 8662 CrossRef CAS PubMed.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS PubMed; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry. Part A: Structure and Mechanisms, Springer, New York, 5th edn, 2007 Search PubMed.
- V. Polo, J. Andrés, S. Berski, L. R. Domingo and B. Silvi, J. Phys. Chem., 2008, 112, 7128 CrossRef CAS PubMed.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, Lett. Org. Chem., 2010, 7, 432 CrossRef CAS; (b) L. R. Domingo and J. A. Sáez, J. Org. Chem., 2011, 76, 373 CrossRef CAS PubMed.
- (a) L. R. Domingo, P. Pérez and J. A. Sáez, Org. Biomol. Chem., 2012, 10, 3841 RSC; (b) L. R. Domingo, P. Pérez and J. A. Sáez, Tetrahedron, 2013, 69, 107 CrossRef CAS PubMed.
- L. R. Domingo, E. Chamorro and P. Pérez, Org. Biomol. Chem., 2010, 8, 5495 CAS.
- (a) P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed; (b) D. H. Ess, G. O. Jones and K. N. Houk, Adv. Synth. Catal., 2006, 348, 2337 CrossRef CAS.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417 CrossRef CAS.
- L. R. Domingo, P. Pérez and D. E. Ortega, J. Org. Chem., 2013, 78, 2462 CrossRef CAS PubMed.
- L. R. Domingo, M. J. Aurell, P. Pérez and J. A. Sáez, RSC Adv., 2012, 2, 1334 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Study of the A-DA reaction between N-aryl imine 13a and 2-methylene-1,3-dioxolane 16. Study of the mechanism of the BF3 catalysed A-DA reaction of N-phenyl-C-ethoxycarbonyl imine 26 with MVE 23. Table with thermodynamic data. ELF topological analysis of bonding change along the A-DA reaction between BF3:imine complex 18 and ER ethylene 16. Cartesian coordinates of the stationary points. See DOI: 10.1039/c4ra02916j |
| This journal is © The Royal Society of Chemistry 2014 |