Preparation of cellulose and pulp carbamates through a reactive dissolution approach†
Sara R. Labafzadeh*,
Jari S. Kavakka,
Kashmira Vyavaharkar,
Katja Sievänen and
Ilkka Kilpeläinen
Laboratory of Organic Chemistry, Department of Chemistry, University of Helsinki, P.O. Box 55, FIN-00014, Helsinki, Finland. E-mail: sara.labaf@helsinki.fi; Tel: +358453290199
First published on 12th May 2014
Abstract
Carbamoylation of various cellulosic materials was systematically studied, using a reactive dissolution approach. Reactions with cellulose, or pulp and aromatic isocyanates, were initiated as heterogeneous mixtures in hot pyridine. As the reaction proceeded, homogeneous solutions were obtained. However, attempts to synthesize highly substituted cellulose carbamates, with aliphatic isocyanates in pyridine, failed as homogeneous solutions were not achieved, even after long reaction times. Consequently, aliphatic cellulose carbamates were prepared via reactive dissolution in dimethylacetamide (DMA), with dibutyltin dilaurate (DBTL) as catalyst. Reactions of cellulose with aliphatic isocyanates in DMA/DBTL at elevated temperature also ended up as homogeneous solutions. The success of the carbamoylation reaction was assessed by means of FTIR, 1H, 13C, 31P and diffusion-ordered spectroscopy (DOSY) NMR. Carbamoylation of pre-hydrolysis hardwood kraft pulp (PHK), hardwood kraft pulp (HKP) and hemicellulose-poor hardwood kraft pulp (HPHKP) were also studied under similar conditions. Highly soluble carbamates were also obtained with the pulp derivatives. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) of the derivatives showed no significant improvement in the thermal properties compared with the untreated cellulose. However, DSC thermograms of some samples showed a phase change, which is attributed to a glass transition temperature (Tg). Also, a two-step decomposition process was observed in the TGA curves for the derivatives, which is attributed to the cleavage of carbamate substituents through a retro-carbamoylation and degradation of the cellulose backbone. Aromatic carbamates of cellulose showed similar molecular weight distributions compared to intact cellulose. However, for aliphatic cellulose carbamates the derivatization led to products with lower molecular weight.
1 Introduction
Cellulose, derived from natural resources such as wood, annual plants and microbes, represents the most abundant renewable and biodegradable polymeric material on earth. In the past century, cellulose has been a key feedstock for the production of chemicals with various properties and applications, due to its low cost and functional versatility. However, this crystalline and rigid homopolymer has not yet reached its full potential in applications due to its essential insolubility in most common solvents, which is attributed to its high crystallinity through an extended and regular intra- and intermolecular hydrogen-bonding network. Chemical modification of the hydroxyl groups, through a reactive dissolution (heterogeneous modification) strategy, allows this obstacle to be overcome and offers considerable opportunities for preparing cellulose-based polymeric materials.Throughout the years, there has been extensive research to design, synthesize and characterize carbamate derivatives of cellulose, with various properties.1–4 In these reports the products were found to be applicable as regenerated cellulose fibres via the CarbaCell process, where isocyanates are generated in situ from urea.5–8 Other applications were as membrane materials,9–11 chiral stationary phases for liquid chromatography,12–15 derivatives used for the characterization of the molar mass distribution of cellulose, by size exclusion chromatography,16–19 carpet cleaners, sponges and in the formation of lyotropic liquid crystals.20 Cellulose carbamates can be produced from the reaction of cellulose with isocyanates either homogenously or in heterogeneous phase. The homogenously modified derivatives show possible regioselectivity and more even distribution in the substitution pattern, compared to the derivatives created under heterogeneous conditions.21,22 A limited number of solvents are known for the carbamoylation of cellulose including N-methylmorpholine-N-oxide–water (NMMO–H2O), NaOH–urea, N,N-dimethylacetamide (DMA)/LiCl and pyridine- or imidazolium-based ionic liquids.14,15,23–26 However, despite considerable efforts concerning homogenous carbamoylation of cellulose, there is still the challenge to find non-toxic, easily handled, recyclable and economically feasible solvents for the dissolution and chemical processing of cellulose. Most of the above-mentioned solvents have not yet been utilized in commercial applications. Up to now, conventional and commercial processes for cellulose functionalization are heterogeneous.27 Heterogeneous reactions allow a number of advantages including the ease of workup procedure, limited depolymerization and most importantly avoidance of the use expensive and toxic solvents. Cellulose can be functionalized with isocyanates in organic solvents such as pyridine and DMA at elevated temperatures (Scheme 1). The reaction can be initiated heterogeneously in pyridine without the use of any additional catalyst and terminated as a homogenous solution after a few hours, depending on the reagent used. However, when DMA is used it is essential to utilize a catalyst in order to expedite the reaction. Dibutyltin dilaurate (DBTL) has been reported as an efficient catalyst for isocyanate-alcohol–water reactions.28–30 These reactions yield highly substituted and highly soluble products. Many papers have been published dealing with the preparation of cellulose carbamates via an isocyanate-pyridine procedure, with different aromatic and aliphatic isocyanates. In these reports polysaccharide substrates such as cellulose, amylase, chitosan and xylan are studied and they are mostly focusing on the chiral and membrane properties of the resulting products.10,31–34 However, the isocyanate-pyridine method has not been systematically screened to obtain high DS cellulose carbamates, using various pulp samples as starting material and different isocyanates as reagent. In addition, the DMA/DBTL medium has been developed by Mormann and coworkers for carbamoylation of various cellulose sources, without the formation of by-products.35 Mormann et al. illustrated that this medium is more efficient than the pyridine method for production of cellulose phenyl- and butylcarbamates. To the best of our knowledge, no more studies have been systematically investigated using this method for the production of technical pulp-derived carbamates, with different isocyanates.
 | ||
| Scheme 1 Carbamoylation of cellulose using a reactive dissolution approach. | ||
One of the major problems in the synthesis of cellulose carbamates is the formation of undesired low molecular weight (MW) by-products, along with the main product. Isocyanates react with water, present in both cellulose and in the solvent, yielding a symmetrical urea from the isocyanate; through decarboxylation of the carbamic acid and subsequent carbamoylation of the primary amine.35 However, the formation of these ureas can be minimized by a careful drying of both the cellulose sample and the solvent. Moreover, low MW carbamates are formed when alcohols are used as non-solvents, for the precipitation of cellulose carbamate, through reaction with excess isocyanate. These carbamates are often insoluble in alcohols and precipitate along with the desired product. A few attempts have been made to limit or eliminate these by-products.19 Wood et al. found that the undesired carbamates can be isolated from cellulose carbamate when methanol is used as precipitation solvent but only 80% of the carbamates are recovered.36 In continuation of Wood's study, Evans et al. stated that optimum precipitation was achieved with 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 methanol–water solution, as the non-solvent.19 In this ratio the formation of low MW by-products is minimized while the recovery of cellulose carbamate is maximized.
:3 methanol–water solution, as the non-solvent.19 In this ratio the formation of low MW by-products is minimized while the recovery of cellulose carbamate is maximized.
In the current study, preparation of cellulose carbamates with various aromatic and aliphatic isocyanates, through reactive dissolution, was systematically investigated. Carbamoylation of cellulose with aromatic isocyanates was conducted in pyridine, while DMA/DBTL was utilized for modification of cellulose with aliphatic isocyanates. This work also provided conditions for optimum precipitation of carbamates without co-precipitation of low MW by-products. Additionally, different types of pulp including pre-hydrolysis hardwood kraft pulp (PHK), hardwood kraft pulp (HKP) and hemicellulose-poor hardwood kraft pulp (HPHKP) were used as raw materials, with microcrystalline cellulose (MCC) as a reference.
2 Experimental
2.1. Materials
PHK from eucalyptus was supplied by Bahia (Brazil) containing 2.5% hemicelluloses with viscosity of 468 ml g−1. HKP, provided by Metsä Fibre-Kaskinen pulp mill (Finland), contained 24.7% hemicelluloses and gave an intrinsic viscosity of 770 ml g−1. HPHKP, provided by the Technical Research Center of Finland (VTT), contained 13.7% hemicelluloses and gave an intrinsic viscosity of 870 ml g−1. PHK and HPHKP were used as received without further treatment, while HKP was fibrillated in hot ethanol and dried in a vacuum oven at 50 °C, in order to maximize the surface area for solvent penetration. Avicel® was used as the MCC sample and was purchased from Sigma-Aldrich. Pyridine and chloroform were provided by VWR international, BDH Prolabo. 1,4-Dioxane was purchased from Labscan, analytical sciences (Switzerland) and was distilled prior to use. All other reagents were purchased from Sigma-Aldrich and were used without further purification.2.2. Synthesis of cellulose carbamates with aromatic isocyanates in pyridine
MCC (0.25 g, 1.54 mmol), pyridine (12.5 ml) and aromatic isocyanate (6 eq. per AGU) were placed in a 20 ml vial. The mixture was heated to 90 °C with stirring under argon. The reaction time was varied depending on the isocyanate (Table 1). When a clear solution was obtained, the product was precipitated by addition into 200 ml of a methanol–water mixture (v/v = 7:3) and rapid stirring. The precipitate was filtered off and washed with 100 ml of methanol–water (v/v = 9:1). The precipitate was then triturated in 100 ml of methanol–water (7/3, v/v) for 1 h, filtered off and then dried under vacuum. Entries 1 and 2 in Table 1 were pure after this stage, while entries 3, 4 and 5 needed further purification. Therefore, they were dissolved in dry dioxane (25 ml), precipitated by addition of methanol (100 ml), were filtered and then dried. The final white products were obtained with weight percent gains (WPGs) of 116 to 200%, depending on the isocyanate.
| No. | Substrate | Isocyanate | Solvent | Reaction time (h) | Yield (%) | DS (% functionalized OH)e | Td(onset) (°C) |
|---|---|---|---|---|---|---|---|
| a Pre-hydrolysis hardwood kraft pulp.b Hardwood kraft pulp.c Hemicellulose-poor hardwood kraft pulp.d Addition of isocyanate was repeated two and four times in intervals of 24 h for MCC and pulps, respectively.e The percentage of the functionalized hydroxyls in the sample, given as % and not as a fraction of 3 (3 OH groups per AGU). | |||||||
| 1 | MCC | 3-(Trifluoromethyl)phenyl | Pyridine | 3 | 50 | 2.9 (99) | 278 |
| 2 | MCC | 4-(Trifluoromethyl)phenyl | Pyridine | 3 | 50 | 3 (100) | 300 |
| 3 | MCC | 2,4-Dimethylphenyl | Pyridine | 24 | 65 | 3 (100) | 243 |
| 4 | MCC | 3,5-Dichlorophenyl | Pyridine | 3 | 54 | 2.9 (99.6) | 325 |
| 5 | MCC | 4-Methoxyphenyl | Pyridine | 24 | 80 | 3 (100) | 289 |
| 6 | MCC | Phenyl isocyanate | Pyridine | 24 | 95 | 2.9 (99.4) | 304 |
| 7 | PHKa | Phenyl isocyanate | Pyridine | 24 | 87 | −(99.4)e | 277 |
| 8 | HPHKPc | Phenyl isocyanate | Pyridine | 24 | 88 | −(99.6)e | 302 |
| 9 | HKPb | Phenyl isocyanate | Pyridine | 24 | 93 | −(99.2)e | 263 |
| 10 | MCC | Cyclohexyl isocyanate | DMA/DBTL | 72d | 71 | 2.6 (96.1) | 256 |
| 11 | PHK | Cyclohexyl isocyanate | DMA/DBTL | 144d | 68 | −(95.4)e | 253 |
| 12 | MCC | Hexyl isocyanate | DMA/DBTL | 24 | 67 | 3 (100) | 233 |
| 13 | MCC | Octyl isocyanate | DMA/DBTL | 24 | 96 | 3 (100) | 240 |
| 14 | MCC | Undecyl isocyanate | DMA/DBTL | 24 | 84 | 3 (100) | 258 |
2.3. Synthesis of cellulose carbamates with aliphatic isocyanates in DMA/DBTL
Carbamoylation of cellulose with aliphatic isocyanates was performed in a mixture of DMA and DBTL, as catalyst. MCC (0.25 g, 1.54 mmol) and DMA (10 ml) were introduced into a 20 ml vial under argon. The resulting suspension was then heated to 90 °C with 6 eq. of isocyanate per AGU in the presence of 0.1 g (0.16 mmol) of DBTL. The mixture was stirred for 1 to 6 days depending on the isocyanate used (see Table 1). The addition of isocyanate was repeated in intervals of 24 h for cyclohexyl derivatives in order to obtain a clear solution. The treated cellulosic materials were then recovered by addition into 200 ml of methanol–water mixture (v/v = 5:1) and rapid stirring. After filtration, the crude solid product was further purified by dissolution into chloroform (25 ml) and precipitation by addition of methanol (100 ml), with rapid stirring. After filtration and drying the products were obtained as white powders with WPGs of 104–292%, depending on the isocyanate used.
2.4. Methods of characterization
IR spectra from solid samples were recorded with a Bruker Alpha ATR-FT-IR spectrometer.1H and 13C NMR spectra were collected using a Varian Unity INOVA 600 spectrometer (600 MHz proton frequency) equipped with a 5 mm direct detection broadband probe head at 27 °C. Diffusion ordered spectroscopy (DOSY) were collected on a Varian Unity INOVA 500 NMR spectrometer (500 MHz proton frequency) equipped with 5 mm triple resonance (1H, 13C and 15N) gradient probe head.
Degrees of substitution (DS) were determined using a 31P NMR method, according to a procedure described previously for chloroform soluble samples.37 For those samples which were insoluble in chloroform, hot pyridine (500 μl) was used for the dissolution of the carbamates: cellulose carbamate (25 mg) was placed in a 10 ml screw-top vial and pyridine (500 μl) was added. The mixture was heated to 80 °C until the sample was fully dissolved. Chloroform-d (1 ml), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP, 200 μl, 1.26 mmol), internal standard solution (e-HNDI, 121.5 mM in pyridine–chloroform-d (3:2 v/v, 125 μl, 0.0152 mmol)) and Cr(acac)3 (1 ml, 0.08 M in chloroform-d) were also added to the solution and agitated until homogeneous. 31P NMR spectra were recorded and DS values were calculated according to previously described equation.37
Gel permeation chromatography (GPC) was used to determine molecular weight distributions (MWDs) of derivatives, with respect to pullulan standards. GPC measurements were performed using an Agilent system including degasser, pump, autosampler, column oven (1100 series), diode array UV detector (1050 series) and refractive index detector (1200 series). The gel permeation column was Agilent PLgel Organic 10 μm MIXED-B (7.5 × 300 mm). LiCl–DMA (0.5% w/v) was used as the mobile phase at a flow rate of 0.5 ml min−1. The Agilent Chemstation (rev. A. 10.02) with Agilent GPC addon (rev. A 02.02) was used to calculate the MWDs.
Thermal properties of cellulose carbamates were analysed by means of TGA and DSC. TGA was recorded using a Mettler-Toledo TGA/SDTA 851e using a temperature range of 50 to 600 °C and a heating rate of 10 °C min−1 in 50 ml min−1 N2. The thermal decomposition temperature (Td) was taken as the onset of significant (≥0.5%) weight loss. Differential scanning calorimetry (DSC) was carried out by DSC Q200. All the experiments were conducted at a heating rate of 10 °C min−1 and cooling rate of 5 °C min−1 in an atmosphere of nitrogen. A heating–cooling–heating cycle was used and after cooling the data from the second heating cycle was analysed. The maximum heating temperature was chosen to be below the thermal decomposition temperature of the carbamate.
Scanning electron microscopy (SEM) imaging of unmodified and modified derivatives were obtained with a Hitachi S-4800 FESEM.
3 Results and discussion
Previously, it was found that pyridine was a suitable reaction media for esterification of cellulosic substrates, using aromatic and long-chain aliphatic acid chlorides, whereas attempts to obtain short-chain (C < 6) and also unsaturated aliphatic cellulose esters failed.38 In continuation of the esterification study, the carbamoylation of different sources of cellulose with a variety of isocyanates in both pyridine and in DMA/DBTL media were investigated. Reactions were initially heterogeneous at high temperature and after a few hours clear homogeneous solutions were obtained, caused by the reactive dissolution of cellulose in the reaction media.3.1. Synthesis of cellulose carbamates
Reactions of cellulose and an aromatic isocyanate were carried out in pyridine at elevated temperatures for different reaction times, depending on the reactivity of isocyanate, to give homogeneous solutions. In the case of aliphatic isocyanates, no clear solutions were obtained. Thus, cellulose carbamates with aliphatic chains were synthesized in DMA/DBTL (Table 1). Longer reaction times (up to 6 days) and regular addition of isocyanates over a period of time (Table 1) were required to give homogeneous solutions of cyclohexylcarbamates.When a homogenous solution was achieved, the resulting products were regenerated with a mixture of methanol–water (v/v= 7:3 for aromatic and 5:1 for aliphatic). Some samples required further purification with a dissolution/precipitation process, using dioxane–methanol for aromatic samples and chloroform/methanol for aliphatic samples. The additional purification method subsequently led to reduced yields (Table 1). No by-products with low MW were present using these purification methods.
Further studies were performed on carbamoylation of various pulp samples, with different hemicellulose contents, in order to investigate both the suitability of cheaper pulp samples as starting materials and whether the reactivity is significantly influenced by the pulp. For this purpose, reactions were performed in pyridine with phenyl isocyanate as reactant. Furthermore, carbamoylation of PHK with cyclohexyl isocyanates were carried out in DMA/DBTL and the results were compared to similar derivatives obtained from MCC (Table 1). In short, highly substituted carbamates were obtained with all pulp samples.
3.2. Infrared analysis of cellulose carbamates
Carbamates of cellulose were characterized by means of ATR-IR to ensure the reaction of hydroxyl groups with isocyanate. The IR spectra of unmodified and modified PHK with phenyl- and cyclohexyl isocyanate are shown in Fig. 1. Compared with the untreated pulp, the new absorption peak at around 1700 cm−1 is observable for modified pulp, which is attributed to the stretching vibration of the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the carbamate group. Another typical absorption band for the carbamoylated cellulose appeared at 3322 cm−1 which is assigned to the (NH) of the carbamate group. In addition, extra peaks at around 1443 cm−1 and 1600 cm−1 correspond to the aryl ring of phenyl isocyanate and at around 2880 cm−1, corresponding to the alkyl chains of cyclohexyl isocyanate. Appearance of the above-mentioned absorptions, along with the elimination of hydroxyl group peak at 3300 cm−1, represent strong evidence for reaction of isocyanates with the pulp samples. The IR spectrum of the resulting HKP and HPHKP, treated with phenyl isocyanate, showed similar peaks and the data is presented in the ESI.†
O) of the carbamate group. Another typical absorption band for the carbamoylated cellulose appeared at 3322 cm−1 which is assigned to the (NH) of the carbamate group. In addition, extra peaks at around 1443 cm−1 and 1600 cm−1 correspond to the aryl ring of phenyl isocyanate and at around 2880 cm−1, corresponding to the alkyl chains of cyclohexyl isocyanate. Appearance of the above-mentioned absorptions, along with the elimination of hydroxyl group peak at 3300 cm−1, represent strong evidence for reaction of isocyanates with the pulp samples. The IR spectrum of the resulting HKP and HPHKP, treated with phenyl isocyanate, showed similar peaks and the data is presented in the ESI.†
 | ||
| Fig. 1 ATR-IR spectra of unmodified PHK (solid), PHK phenyl carbamate 7 (dots) and PHK cyclohexyl carbamate 11 (dashes). | ||
3.3. NMR analysis of cellulose carbamates
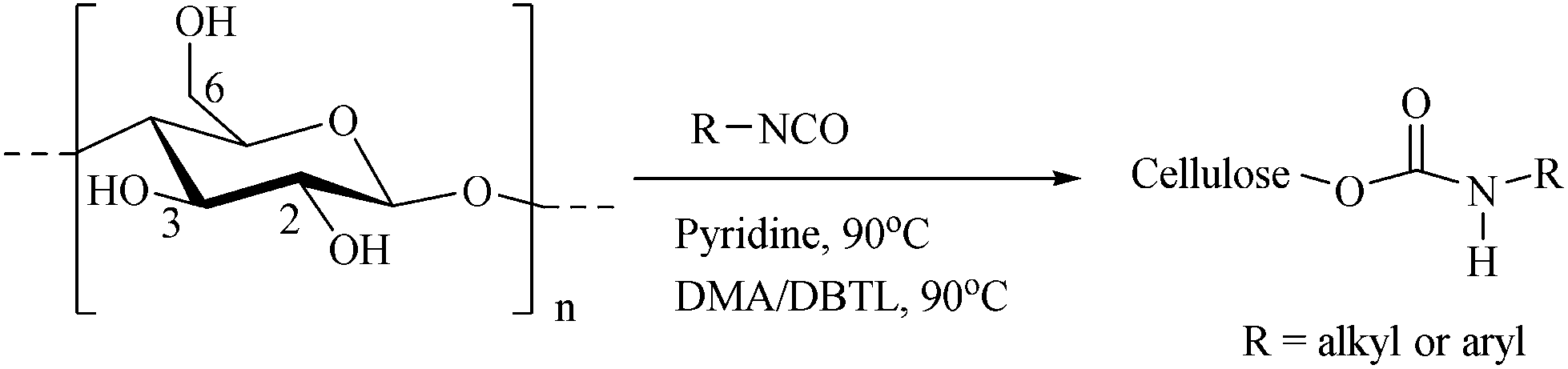
Besides IR analysis, chemical structure and purity of the carbamoylated cellulose derivatives was also studied by NMR spectroscopy. Fig. 2 and 3 illustrate the 1H and 13C NMR spectra of carbamoylated PHK, for phenyl-(entry 7, Table 1) and cyclohexyl isocyanate (entry 11, Table 1), respectively. Resonance assignments are also shown. The NMR spectra of other cellulose carbamates are presented in ESI.†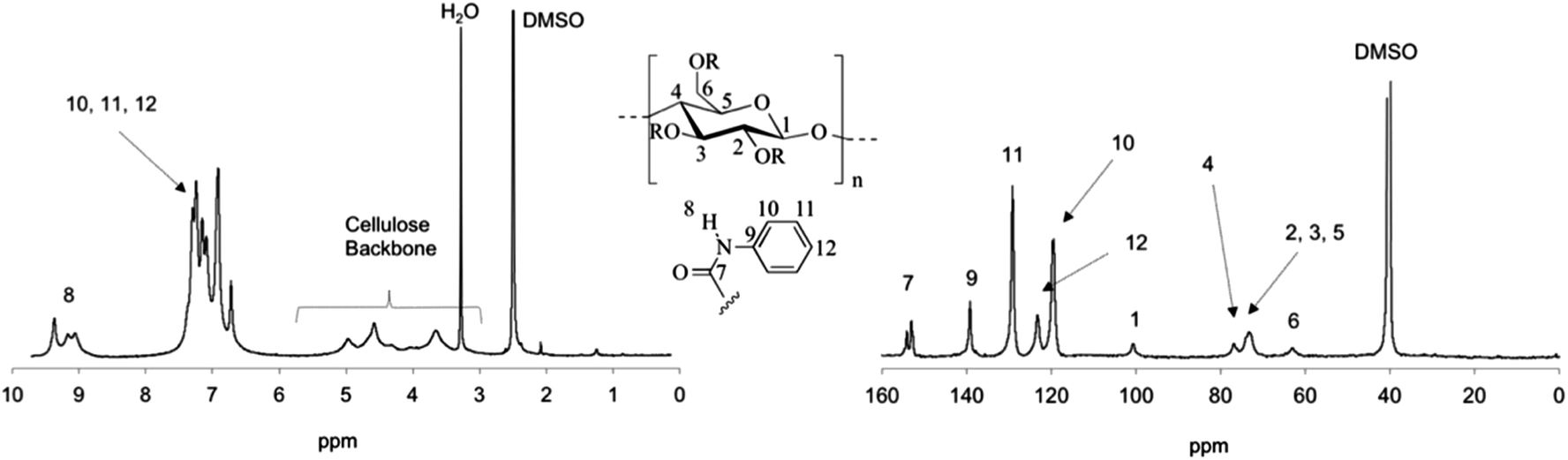 | ||
| Fig. 2 1H NMR (left) and 13C NMR (right) spectra of PHK phenylcarbamate 7 in DMSO-d6. | ||
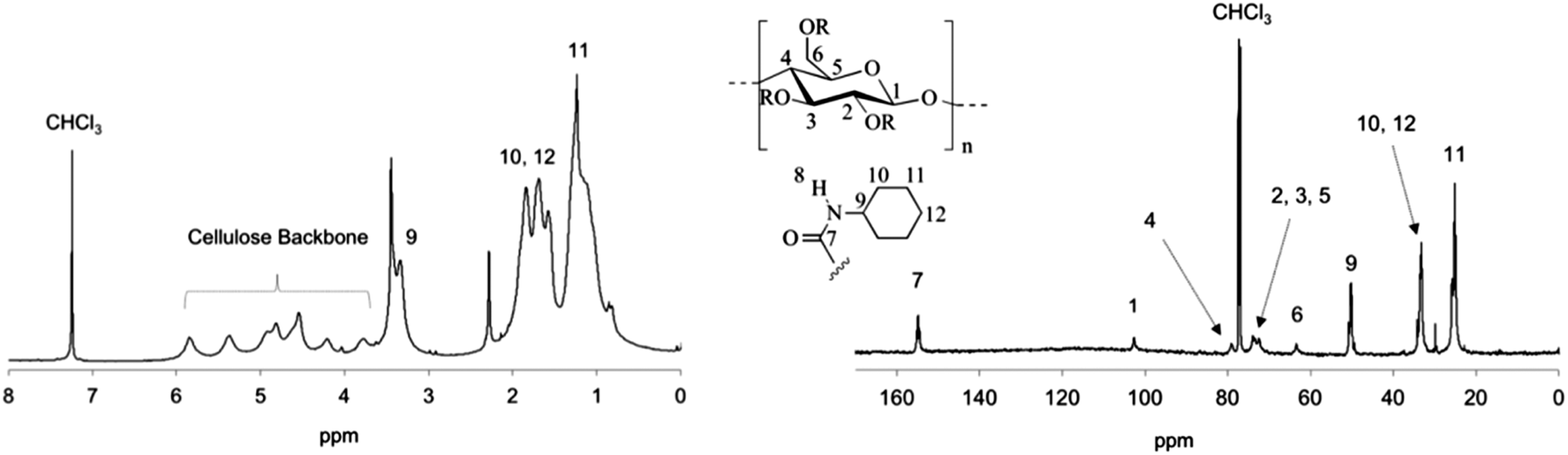 | ||
| Fig. 3 1H NMR (left) and 13C NMR (right) spectra of PHK cyclohexylcarbamate 11 in chloroform-d. | ||
Low MW by-products such as methyl cyclohexylcarbamate are formed when methanol is used to quench the reaction and precipitate the products. These impurities are often impossible to distinguish from functionalized biopolymer using conventional 1H and 13C NMR spectroscopy. Therefore, diffusion-ordered spectroscopy (DOSY) NMR was used to resolve any small molecules formed during the reaction or precipitation. DOSY measures molecular mobilities i.e. diffusion rates in an NMR tube. The diffusion coefficient (D) of a molecule is proportional to its hydrodynamic radius. Therefore, DOSY offers a way to resolve different compounds in a mixture based on differences in the size and shape of the molecule. The DOSY spectrum of PHK cyclohexylcarbamate (Fig. 4) shows a small peak at 2.4 ppm which might correspond to impurities, although diffusion is slower than for methanol (signal appearing at 3.5 ppm) indicating the absence of further low MW species.
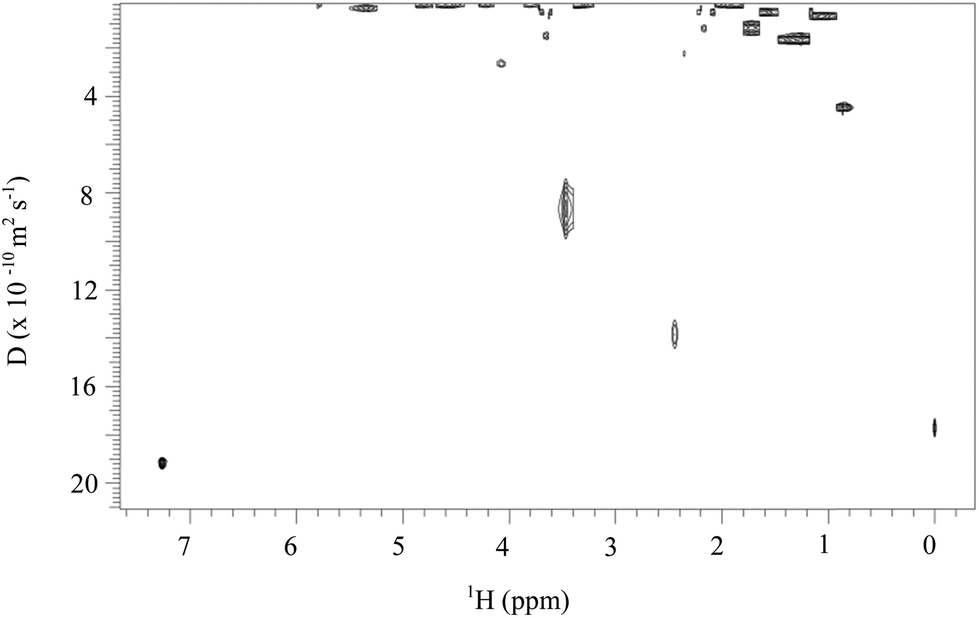 | ||
| Fig. 4 DOSY NMR spectrum of PHK cyclohexylcarbamate 11 in chloroform-d. | ||
3.4. 31P NMR analysis for DS determination of cellulose carbamates
Cellulose carbamates with high DS values were obtained using both pyridine and DMA/DBTL. The DS values for the derivatives were again measured using the 31P NMR method.37 The data presented in Table 1 shows that the DS close to 3 (100%) were achieved for all derivatives. As pulps also containing hemicelluloses, the percentage of the functionalized hydroxyl groups in the sample were calculated for modified pulps, as the traditional concept of DS (3 OH per AGU) cannot apply. Comparing the functionalized hydroxyls of phenyl isocyanate treated pulps (entry 7, 8 and 9, Table 1), it can be also concluded that the presence of hemicelluloses in the pulp and the pulps inherent reactivity do not have any significant effect on the outcome of these reactions.3.5. Gel permeation chromatography of cellulose carbamates
The molecular weight distributions (MWDs) of the obtained cellulose carbamates were studied by the means of GPC. The data shown in Fig. 5 illustrates that carbamoylation of MCC, with aromatic isocyanates in pyridine, does not influence the MWDs significantly, from the starting MCC. Similarly, aliphatic carbamoylation of cellulose with hexyl isocyanate in DMA/DBTL does not significantly change the MWDs of the starting cellulose. The derivatization of MCC with either octyl or cyclohexyl isocyanates, however, resulted in a shift of the MWDs to the lower molecular weights (Fig. 6). Additionally, the phenyl carbamoylation of the PHK and HKP did not affect the MWDs. PHK cyclohexylcarbamate showed a broader molar mass distribution. In case of the HPHKP, however, carbamoylation introduced major changes in the MWDs and resulted in a reduction of the molecular weight, compared with that of the untreated HPHKP (data shown in ESI†). | ||
| Fig. 5 MWDs of MCC (solid), MCC 3-(trifluoromethyl)phenylcarbamate 1 (dots), MCC 2,4-dimethylphenylcarbamate 3 (short dashes), MCC 4-methoxyphenylcarbamate 5 (long dashes) and MCC phenyl carbamate 6 (dash-dot-dot). | ||
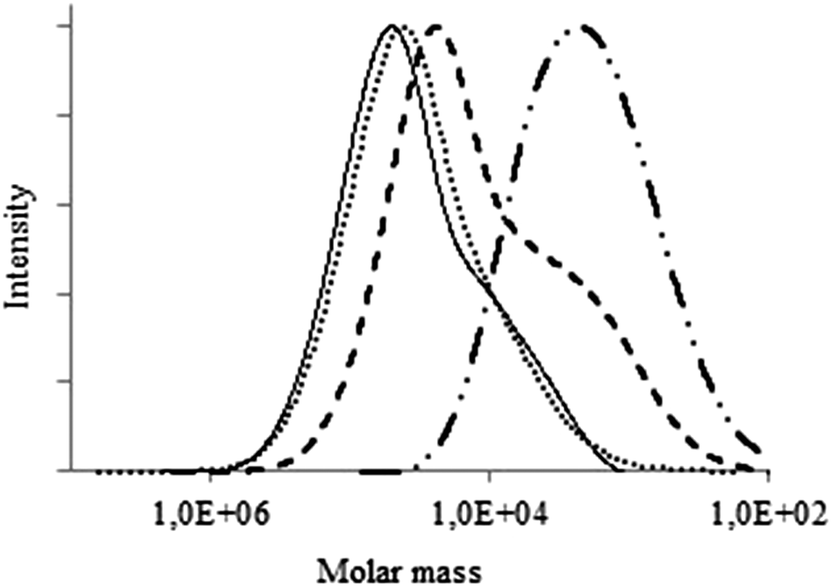 | ||
| Fig. 6 MWDs of MCC (solid), MCC hexylcarbamate (dots), MCC cyclohexyl carbamate (dashes) and MCC octylcarbamate (dash-dot-dot). | ||
The presence of low MW by-products could also be observed by appearance of an additional sharp peak in the MWD of derivatives. Observations from GPC confirm the absence of by-products for all derivatives, in agreement with the results from the 1H, 13C and DOSY NMR spectra.
3.6. Thermal properties of cellulose carbamates
The thermal properties of the derivatives were investigated by means of TGA and DSC. The thermal stability of the modified celluloses varies with the structure of the modifying reagent. Decomposition temperatures (Td) of all the derivatives are summarized in Table 1. It was found that Td of cellulose carbamates are lower than for intact MCC and pulps (around 320 °C and 328 °C, respectively). This shows that carbamoylation of cellulose does not improve the thermal stability of cellulose. Thermal stability of cellulose and cellulose derivatives are mainly influenced by their crystallinity.39 Therefore, the decrease in decomposition temperatures may be partly due to the loss of crystallinity afforded by carbamoylation, which is thermally degraded faster than crystalline regions.40 However, cellulose dichlorophenylcarbamate 4 is an exception with Td of 325 °C, which is higher than for intact MCC (320 °C). TGA curves for PHK treated with phenyl isocyanate and cyclohexyl isocyanate are shown in Fig. 7. In addition to an initial loss of moisture at around 100 °C, a major weight loss attributed to decomposition of cellulose occurs. This may include a mechanism which involves elimination of the starting isocyanate, as carbamate groups are thermally labile.40 In order to prove the elimination of carbamate group under thermal conditions, simultaneous differential thermal analysis (SDTA) results along with IR were used. SDTA curves of phenyl carbamoylated cellulose derivatives show two endotherms, which are assigned to elimination of phenyl isocyanate (lower temperature) and decomposition of cellulose (higher temperature) (Fig. 8). Cellulose phenylcarbamate 6 was heated up to 346 °C, which is under the decomposition temperature for cellulose (designated by * at the Fig. 8, solid thermogram). The residual materials were further characterized by means of ATR-IR spectroscopy. Cleavage of elimination of the isocyanate can be confirmed by the absence of absorption peaks at 1700 cm−1 and 3322 cm−1 and also by appearance of large hydroxyl groups at 3300 cm−1 (Fig. 9).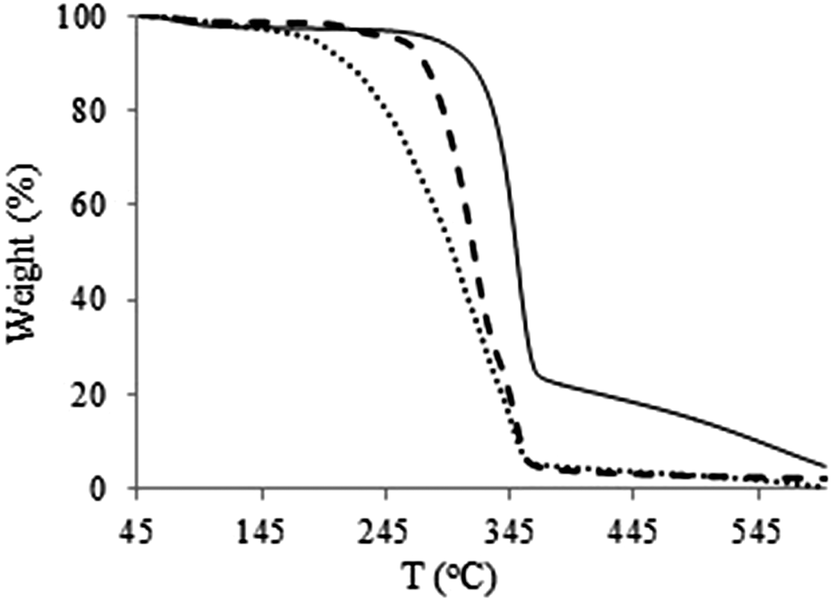 | ||
| Fig. 7 TGA curves of unmodified PHK (solid), PHK cyclohexylcarbamate 11 (dots) and PHK phenylcarbamate 7 (dashes). | ||
 | ||
| Fig. 8 SDTA curves of MCC phenylcarbamate 6 (solid), PHK phenylcarbamate 7 (dots), HPHKP phenylcarbamate 8 (dashes) and HKP phenyl carbamate 9 (dash-dot-dot). | ||
 | ||
| Fig. 9 ATR-IR spectra of unmodified MCC (solid) and MCC phenyl carbamate after thermal treatment with TGA up to 346 °C (Fig. 8, solid thermogram, at point designated by *) (dots). | ||
Phenyl isocyanate treated PHK shows two steps in the TGA (Fig. 7) and STDA (Fig. 8) curves. This might be due to differences in the thermal stability of the carbamate functionalities derived from primary and secondary hydroxyl groups. Furthermore, the additional step in the TGA traces have no developed plateau, due to the low thermal stability of the residual cellulose after elimination of the isocyanates. Cyclohexylcarbamoylated PHK showed similar trends (Fig. 7 and 8). Its decomposition, nevertheless, was found to be more gentle than cellulose treated with phenyl isocyanate.
DSC analysis was used to investigate phase changes in the materials. No notable transitions were observed for the derivatives (data shown in ESI†). Therefore, it can be concluded that the treatment of cellulose with the inexpensive phenyl and cyclohexyl isocyanates has no considerable effect on the thermal properties of the products. There are, however, some exceptions. DSC thermograms of samples 1, 3 & 4 showed phase changes at 199 °C, 192 °C and 172 °C, respectively, which may be attributed to the glass transition temperature (Tg) (Fig. 10).
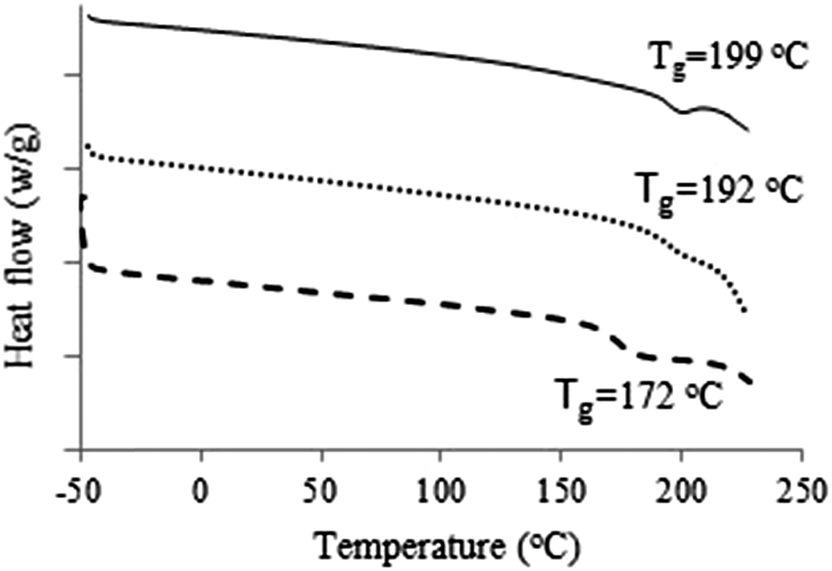 | ||
| Fig. 10 DSC analysis of MCC 3-(trifluoromethyl)phenylcarbamate 1 (solid), MCC 2,4-dimethylphenylcarbamate 3 (dots) and MCC 3,5-dichlorophenylcarbamate 4 (dashes). | ||
3.7. Morphology of cellulose carbamates
Fibre morphology can significantly affect the properties of cellulose derivatives and their subsequent applications. Scanning electron microscopy (SEM) was used to examine the morphology of the derivatives and to attain a better understanding of the effects of the attached carbamate group to cellulose surface. Fig. 11 shows the SEM photographs of the intact PHK and treated PHK with different modifying reagents. A change in the surface morphology of the derivatives is notable compared to untreated pulp. The fibrous structure after the carbamoylation is completely removed indicating homogeneous modification has taken place, rather than simple swelling of the fibres during the reaction. | ||
| Fig. 11 SEM images of unmodified PHK (left), PHK phenyl carbamate 7 (middle) and PHK cyclohexyl carbamate 11 (right). | ||
4 Conclusions
Synthesis of highly substituted and highly soluble cellulose carbamates were systematically screened through a reactive dissolution approach. Pyridine is an efficient reaction medium yielding cellulose derivatives of high DS, when aromatic isocyanates are used as reactants. It also turned out that soluble aliphatic cellulose carbamates with high DS can be prepared using DMA/DBTL. A substantially longer reaction time (up to 6 days) and addition of isocyanates at intervals was required to attain homogeneous solutions of aliphatic isocyanates. The purification procedure allowed the synthesis of pure carbamates, without presence of any low molecular weight by-products. The reactive dissolution approach also allowed the production of highly substituted cellulose carbamates, even when different technical pulps, with different hemicelluloses contents, were used as starting material.It was demonstrated that there was no improvement in thermal properties of the carbamate derivatives except for samples 1, 3 & 4, which showed glass transition temperatures at 170–200 °C. During thermal decomposition, isocyanates can be eliminated from the carbamate functionalities to give the starting cellulose. The treatment of pulps with aromatic isocyanates in pyridine did not influence the molar mass distribution significantly. However, derivatization with aliphatic isocyanates in DMA/DBTL caused a pronounced degradation of the polymer chains. Cellulose hexylcarbamate was the exception with no significant degradation of cellulose backbone observed.
Acknowledgements
This work was supported by Finnish Bioeconomy Cluster FIBIC Oy as a part of the Future Biorefinery (FuBio) project, by the Academy of Finland and by Biomass Refining Graduate School (BIOREGS) (grants 122534 and 132150). The authors would like to thank Dr Alistair King for his help and productive discussions.References
- M. L. Hassan, C. N. Moorefield and G. R. Newkome, Macromol. Rapid Commun., 2004, 25, 1999–2002 CrossRef CAS
.
- X. Shen, N. Sugie, Q. Duan, Y. Kitajyo, T. Satoh and T. Kakuchi, Polym. Bull., 2005, 55, 317–322 CrossRef CAS
- C. Yin and X. Shen, Eur. Polym. J., 2007, 43, 2111–2116 CrossRef CAS PubMed
- J. Gironès, M. T. B. Pimenta, F. Vilaseca, A. J. F. Carvalho, P. Mutjé and A. A. S. Curvelo, Carbohydr. Polym., 2008, 74, 106–113 CrossRef PubMed
- Comprehensive cellulose chemistry, Functionalization of cellulose, D. Klemm, B. Philipp, T. Heinze, U. Heinze and W. Wagenknecht, Wiley-VCH, Weinheim, 1998, vol. 2, pp. 161–164 Search PubMed.
- Y. Guo, J. Zhou, Y. Song and L. Zhang, Macromol. Rapid Commun., 2009, 30, 1504–1508 CrossRef CAS PubMed
- Y. Guo, J. Zhou, Y. Wang, L. Zhang and X. Lin, Cellulose, 2010, 17, 1115–1125 CrossRef CAS
- L. T. T. Vo, B. Široká, A. P. Manian and T. Bechtold, Carbohydr. Polym., 2010, 82, 1191–1197 CrossRef CAS PubMed
- C. M. Heard and R. Suedee, Int. J. Pharm., 1996, 139, 15–23 CrossRef CAS
- M. Diamantoglou, J. Platz and J. Vienken, Artif. Organs, 1999, 23, 15–22 CrossRef CAS
- F. Z. Khan, M. Shiotsuki, Y. Nishio and T. Masuda, J. Membr. Sci., 2008, 312, 207–216 CrossRef CAS PubMed
- T. Kubota, C. Yamamoto and Y. Okamoto, Chirality, 2003, 15, 77–82 CrossRef CAS PubMed
- X. Chen, C. Yamamoto and Y. Okamoto, Pure Appl. Chem., 2007, 79, 1561–1573 CrossRef CAS
- European Pat., EP0157365, Y. Okamoto and K. Hatada, 1958
- US Pat., US5589061, J. W. Russel, 1996
- F. Berthold, K. Gustafsson, R. Berggren, E. Sjöholm and M. Lindström, J. Appl. Polym. Sci., 2004, 94, 424–431 CrossRef CAS
- E. Sjöholm, in Handbook Of Size Exclusion Chromatography And Related Techniques, CRC Press, 2003 Search PubMed
- Cellulose degradation in pulp fibers studied as changes in molar mass distributions, R. Berggren, Royal Institute of Technology, 2003 Search PubMed.
- R. Evans, R. H. Wearne and A. F. A. Wallis, J. Appl. Polym. Sci., 1989, 37, 3291–3303 CrossRef CAS
- M. Siekmeyer and P. Zugenmaier, Die Makromolekulare Chemie, 1990, 191, 1177–1196 CrossRef CAS
- K. J. Edgar, T. J. Pecorini and W. G. Glasser, ACS Symposium Series 688, 1998 Search PubMed
- S. Barthel and T. Heinze, Green Chem., 2006, 8, 301–306 RSC
- H. Jin, C. Zha and L. Gu, Carbohydr. Res., 2007, 342, 851–858 CrossRef CAS PubMed
- M. Terbojevich, A. Cosani, M. Camilot and B. Focher, J. Appl. Polym. Sci., 1995, 55, 1663–1671 CrossRef CAS
- S. L. Williamson and C. L. McCormick, J. Macromol. Sci., Part A: Pure Appl.Chem., 1998, 35, 1915–1927 CrossRef
- A. S. Amarasekara and O. S. Owereh, Carbohydr. Polym., 2009, 78, 635–638 CrossRef CAS PubMed
- S. Fischer, K. Thümmler, B. Volkert, K. Hettrich, I. Schmidt and K. Fischer, Macromol. Symp., 2008, 262, 89–96 CrossRef CAS
- G. Engelmann, E. Bonatz, I. Bechthold and G. Rafler, Starch/Staerke, 2001, 53, 560–569 CrossRef CAS
- H. Ni, H. A. Nash, J. G. Worden and M. D. Soucek, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1677–1688 CrossRef CAS
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2012, 113, 80–118 CrossRef PubMed
- W. M. Hearon, G. D. Hiatt and C. R. Fordyce, J. Am. Chem. Soc., 1943, 65, 829–833 CrossRef CAS
- Y. Okamoto, M. Kawashima and K. Hatada, J. Am. Chem. Soc., 1984, 106, 5357–5359 CrossRef CAS
- T. Kubota, C. Yamamoto and Y. Okamoto, Chirality, 2002, 14, 372–376 CrossRef CAS PubMed
- S. H. Kwon, Y. Okamoto, C. Yamamoto, W. Cheong, M. Moon and J. H. Park, Anal. Sci., 2006, 22, 1525–1529 CrossRef CAS
- W. Mormann and U. Michel, Carbohydr. Polym., 2002, 50, 201–208 CrossRef CAS
- B. F. Wood, A. H. Conner and C. G. Hill, J. Appl. Polym. Sci., 1986, 32, 3703–3712 CrossRef CAS
- A. W. T. King, J. Jalomaki, M. Granstrom, D. S. Argyropoulos, S. Heikkinen and I. Kilpelainen, Anal. Methods, 2010, 2, 1499–1505 RSC
- S. Labafzadeh, J. Kavakka, K. Sievänen, J. Asikkala and I. Kilpeläinen, Cellulose, 2012, 19, 1295–1304 CrossRef CAS
- C. Yin, J. Li, Q. Xu, Q. Peng, Y. Liu and X. Shen, Carbohydr. Polym., 2007, 67, 147–154 CrossRef CAS PubMed
- A.-A. M. A. Nada, S. Kamel and M. El-Sakhawy, Polym. Degrad. Stab., 2000, 70, 347–355 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data and thermal analysis results of the prepared cellulose and pulp derivatives are presented. See DOI: 10.1039/c4ra02316a |
| This journal is © The Royal Society of Chemistry 2014 |