 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereospecific alkylation of substituted adenines by the Mitsunobu coupling reaction under microwave-assisted conditions†
M. E. García-Rubiñoa,
M. C. Núñez-Carreteroa,
D. Choquesillo-Lazarteb,
J. M. García-Ruizb,
Yolanda Madridc and
J. M. Campos*a
aDepartamento de Química Farmacéutica y Orgánica, Facultad de Farmacia, c/ Campus de Cartuja s/n, 18071 Granada, Spain. E-mail: jmcampos@ugr.es; Fax: +34 958 243845; Tel: +34 958 243850
bLaboratorio de Estudios Cristalográficos, IACT, CSIC-Universidad de Granada, Avda. de las Palmeras 4, 18100 Armilla, Granada, Spain
cCentro de Instrumentación Científica, Universidad de Granada, Edificio Mecenas, Campus Universitario de Fuente Nueva, 18071 Granada, Spain
First published on 13th May 2014
Abstract
A novel and efficient synthetic method has been developed for the preparation of alkylated aminopurines (N,N-dimethyl-, 2-chloro-N-methyl-, and N-methyladenines) with achiral and chiral 3,4-dihydro-2H-1,5-benzoxathiepin-3-ol by the Mitsunobu reaction under microwave-assisted conditions. This reaction reveals a complete inversion of the stereogenic centre of the secondary alcohol giving an alkylated purine linked to a homochiral six-membered ring. Fifty novel purine derivatives have been prepared. Alkylation sites have been determined by 2D NMR techniques and for three compounds have been confirmed by X-ray crystallography. The N-9/N-3 regioselectivity can be justified by the electronic effects of the substituents at positions 2 and 6 of the purine.
Introduction
In the search for new compounds of therapeutic interest, purine derivatives are very important for their possible interaction with many biological targets. We have reported the fast microwave-assisted synthesis of a series of (RS)-6-substituted-7 or 9-(2,3-dihydro-5H-1,4-benzodioxepin-3-yl)-7H or -9H-purines, with the advantage of the selective preparation of the N-7 or N-9 regioisomers. Our interest in the topic lies in the fact that pyrimidines and purine alkylated products seem to be attractive structures endowed with anti-proliferative properties.1–6The essential biological functions of N-substituted adenine compounds have naturally led to extensive interest in the synthesis of a wide variety of alkylated purine systems as potential analogues or antagonists of naturally occurring adenine derivatives.7 Direct alkylation of adenine, of its simple derivatives, and of its metal salts is the dominant synthetic route to N-substituted adenines, largely because of the convenience and simplicity of such reactions.
There are reports dealing with the alkylation reaction of adenine under the Mitsunobu conditions.8–11 Continuing our studies on multident heterocyclic nucleophiles, we examined the alkylation of adenine using a variety of alkylating agents.12,13 The wide variety of alkylation conditions involved in the literature reports makes it difficult to discern what influence the structural features of the alkylating agent has on the alkylation site. To overcome this problem we have used a typical and standardized alkylation condition to study the reaction of several substituted adenines with racemic 3,4-dihydro-2H-1,5-benzoxathiepin-3-ol, and its two enantiomers through the Mitsunobu reaction under microwave conditions. We have described a series of eleven 2- and 6-substituted (RS)-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-purine derivatives (1–11) (Fig. 1), by applying a standard Mitsunobu protocol that led to a six-membered ring contraction from (RS)-3,4-dihydro-2H-1,5-benzoxathiepin-3-ol via an episulfonium intermediate.14
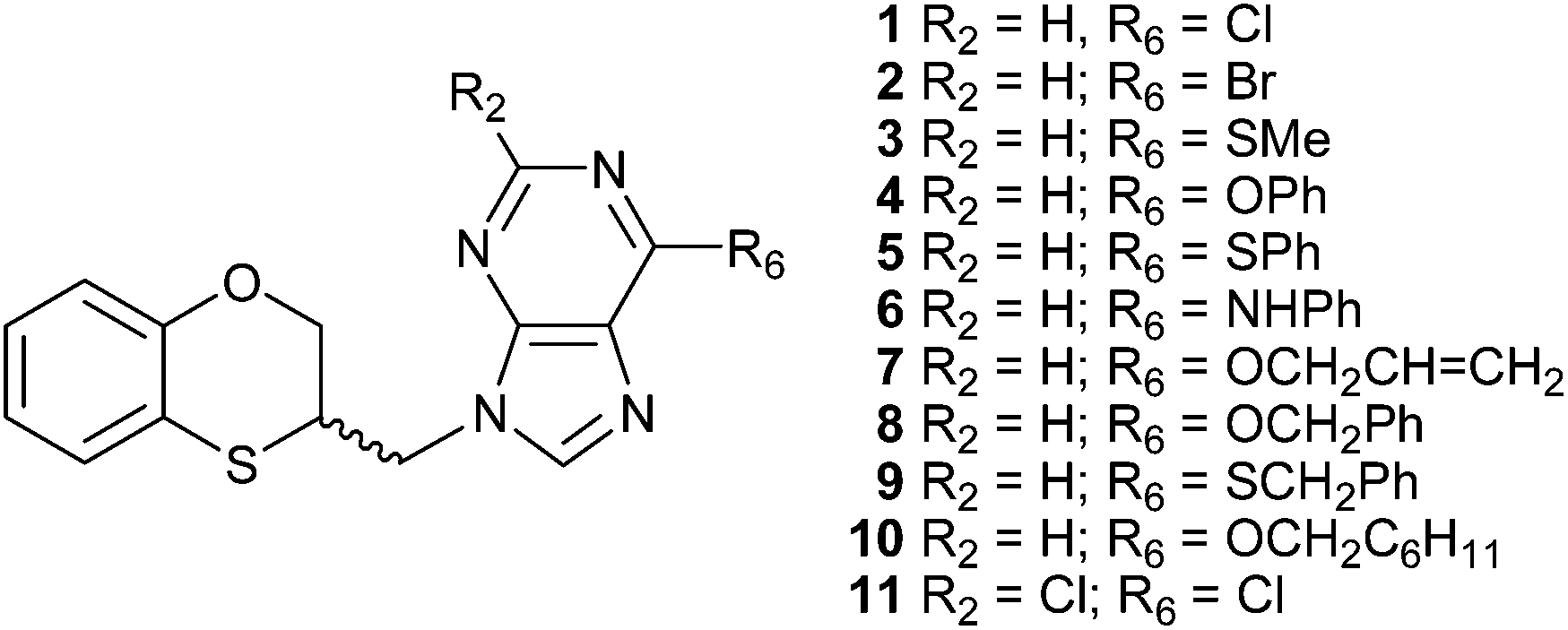 | ||
| Fig. 1 2- and 6-substituted (RS)-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-purine derivatives obtained by the Mitsunobu reaction. | ||
We wish to report herein the reactivity of the racemic and enantiomers of 3,4-dihydro-2H-1,5-benzoxathiepin-3-ol (12) and several adenines, such as N,N-dimethyladenine, 2-chloro-N-methyladenine, and N-methyladenine. The results will be interpreted in terms of multident nucleophile reactivity and SN2 transition state structures for the alkylation reactions. These concepts should allow more confident prediction of the effects that variations in structural features and conditions will have on the alkylation pattern of adenine derivatives during the varied syntheses of alkylated purines.
Results and discussion
Synthesis of (RS)-, (S)- and (R)-3,4-dihydro-2H-1,5-benzoxathiepin-3-ol (12), and (RS)-, (S)- and (R)-2,3-dihydro-1,4-benzoxathiin-2-methanol (13)
Three-carbon (C-3) epoxides bearing halide substituents are highly versatile synthetic building blocks because each carbon is functionalized and a potential site of nucleophilic attack. Epichlorohydrin, in particular, is a readily available C-3 unit that is widely employed in organic and polymer synthesis.15 Compound (RS)-, (S)- and (R)-14 were synthesized under microwave irradiation, racemic or enantiomeric epichlorohydrin as appropriate, 140 °C, 10 min. Both (RS)- and (S)-14 were obtained with a 90% yield, (Scheme 1). Compound (R)-14 was not isolated to obtain (R)-12 and (S)-13 (Scheme 2).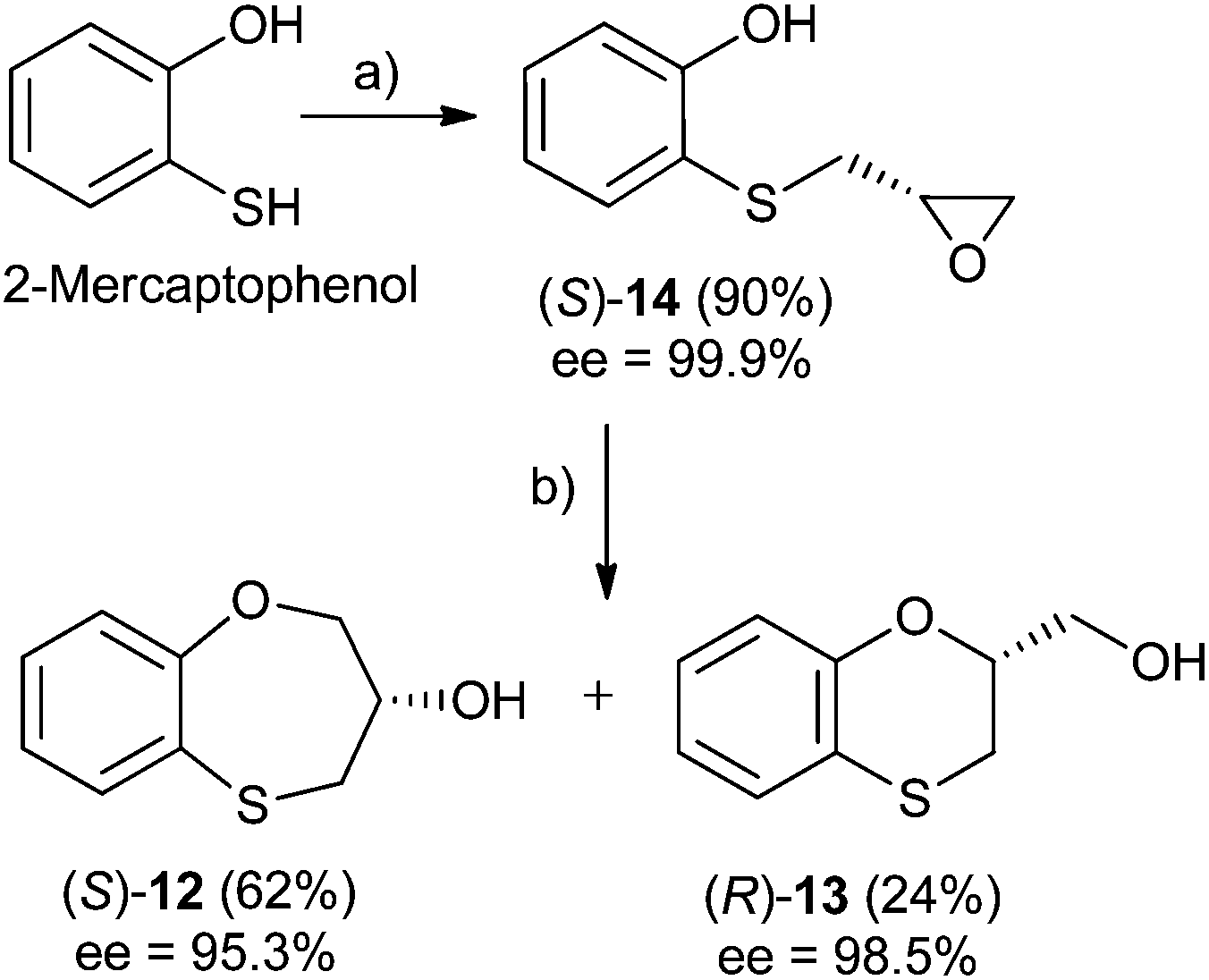 | ||
| Scheme 1 Reagents and conditions: (a) (R)-epichlorohydrin, pyridine, MW, 140 °C, 10 min, DMF; (b) NaOH, H2O, 100 °C, 24 h. | ||
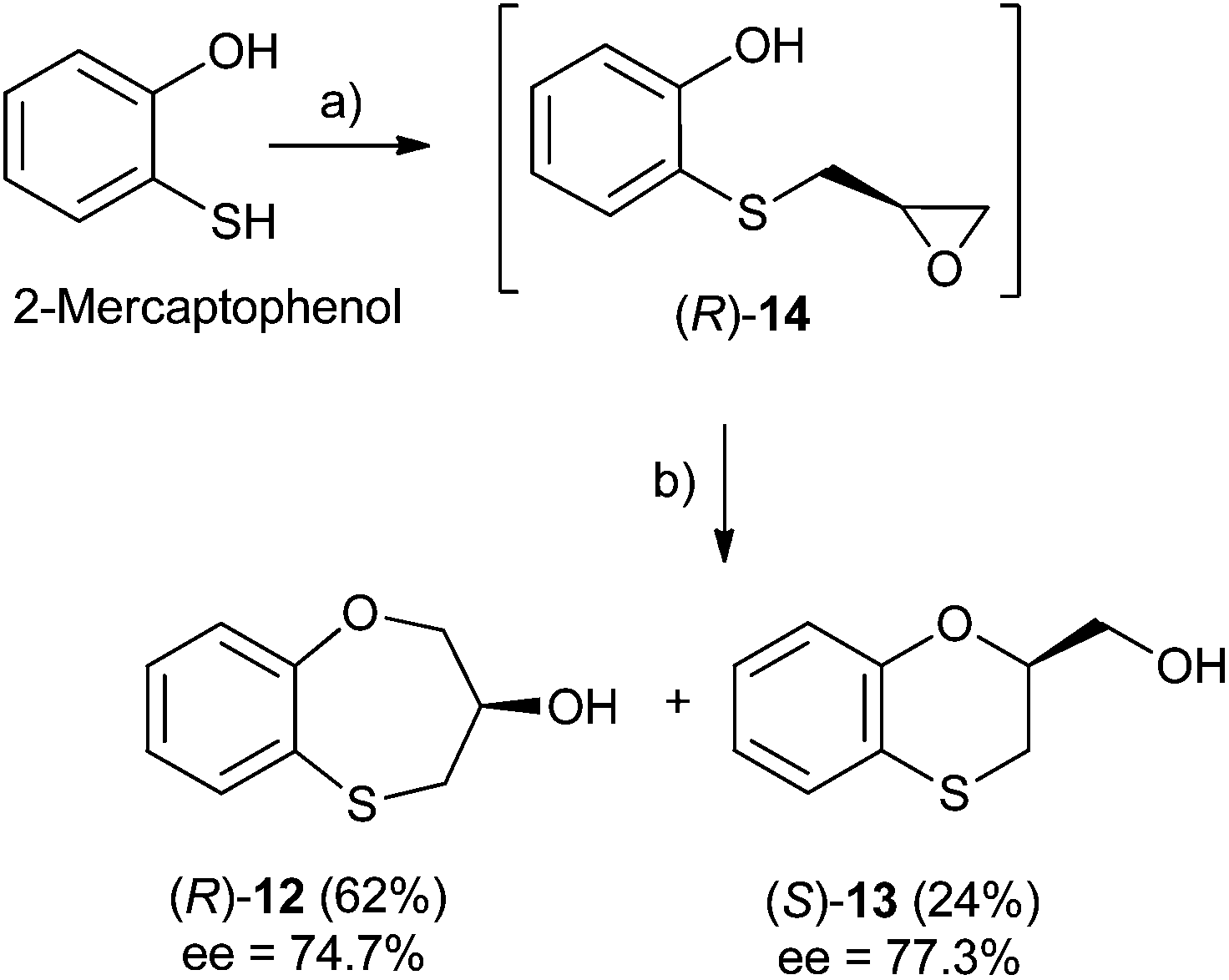 | ||
| Scheme 2 Reagents and conditions: (a) (S)-epichlorohydrin, pyridine, MW, 140 °C, 10 min, DMF; (b) without isolating (R)-14, NaOH, DMF, 100 °C, 24 h. | ||
When 2-mercaptophenol is reacted with (R)-epichlorohydrin, (S)-14 is obtained (90%) with an ee = 99.9% with complete inversion of the stereocentre of its oxirane moiety.16 This implies a complete regioselective process as a consequence of the nucleophilic attack of the sulfanyl anion at the less hindered face of the oxirane moiety of (R)-epichlorohydrin, thus resulting in (S)-14 with an excellent ee. After removing DMF in vacuo and purification by flash column chromatography of the reaction crude, heating (S)-14 with NaOH in H2O at 100 °C gives the cyclization products, (S)-12 (62% yield, ee = 95.3%) and the six-membered primary alcohol (R)-13 (24% yield, ee = 98.5%) (Scheme 1).
Scheme 2 shows the different stereochemical behaviour when the experimental workup was modified using (S)-epichlorohydrin. Although pathway (a) is exactly the same as that shown in Scheme 1, compound (R)-14 is not isolated and NaOH and more DMF are added (instead of water) and the resulting solution is heated at 100 °C for 24 h. After removing DMF and subsequent flash chromatography, (R)-12 (62%, ee = 74.7%) and (S)-13 (24%, ee = 77.3%) are obtained.
The interpretation for the formation of (R)-12 [and for (S)-13 would be very similar] with a marked erosion of its optical purity in relation to the method outlined in Scheme 2 could be rationalized in Scheme 3. The formation of (R)-14 follows the same pattern for (S)-14. As (S)-14 was obtained with a high yield (90%), we decided neither to purify nor isolate (R)-14; although the reaction workup was reduced, the ee worsened due to the presence of HCl in the reaction medium.17 Accordingly, (R)-14 follows two different pathways, i.e., a preferential SN2 process [presumably with an excellent ee, see Scheme 1 with (R)-epichlorohydrin], and a SN1 process with concomitant racemization. Finally, cyclization of pure (R)-14 [route (a) of Scheme 3] and (S)-14 with NaOH at 100 °C leads to (R)-12 and (S)-12, respectively, the enantiomer (R)-12 being produced mainly through pathway (b) and to a lesser extent via pathway (a), whilst (S)-12 is formed only through pathway (a) (Scheme 3).
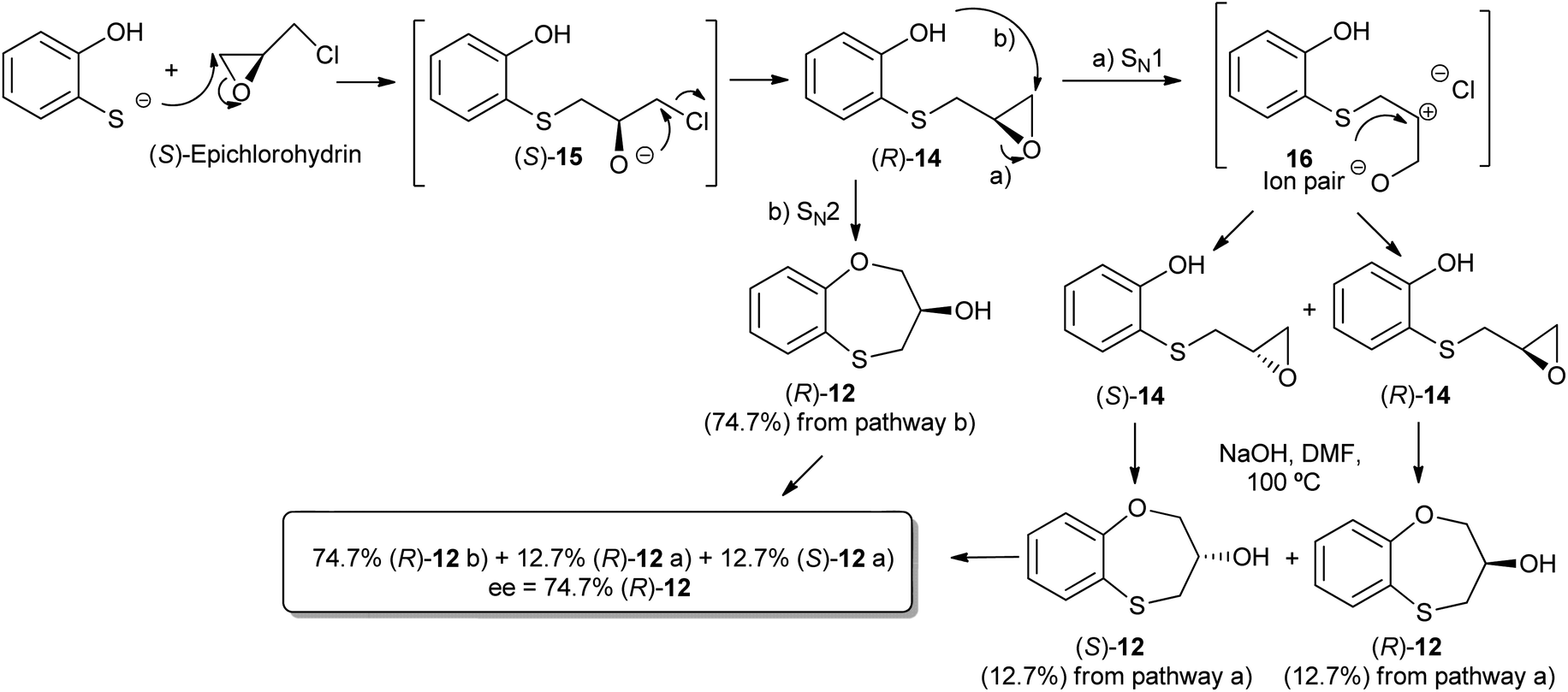 | ||
| Scheme 3 Starting from (S)-epichlorohydrin tentative explanation for the formation of both (R)-12 major and (S)-12 minor compounds, without isolating (S)-15. Formation of the minor (S)-13 compound is not included for simplicity, although the mechanism is similar. | ||
Due to fact that the syntheses of (R)-14 and (S)-14 are carried out under the same conditions, the racemization process is not produced at this stage [confirmed by isolation of (S)-14] and hence, racemization takes place in the following cyclization step, which differs in both enantiomers (Scheme 3).
Reactivity between racemic and enantiomers of 4-dihydro-2H-1,5-benzoxathiepin-3-ol and 6-(N,N-dimethyl)adenine
Scheme 4 shows the reaction pattern between (RS)-12 and N,N-dimethyladenine under microwave irradiation, the N-3 alkylated purine (RS)-17 (32%) being the major compound, and the minor one the N-9 alkylated purine (RS)-18 (5%).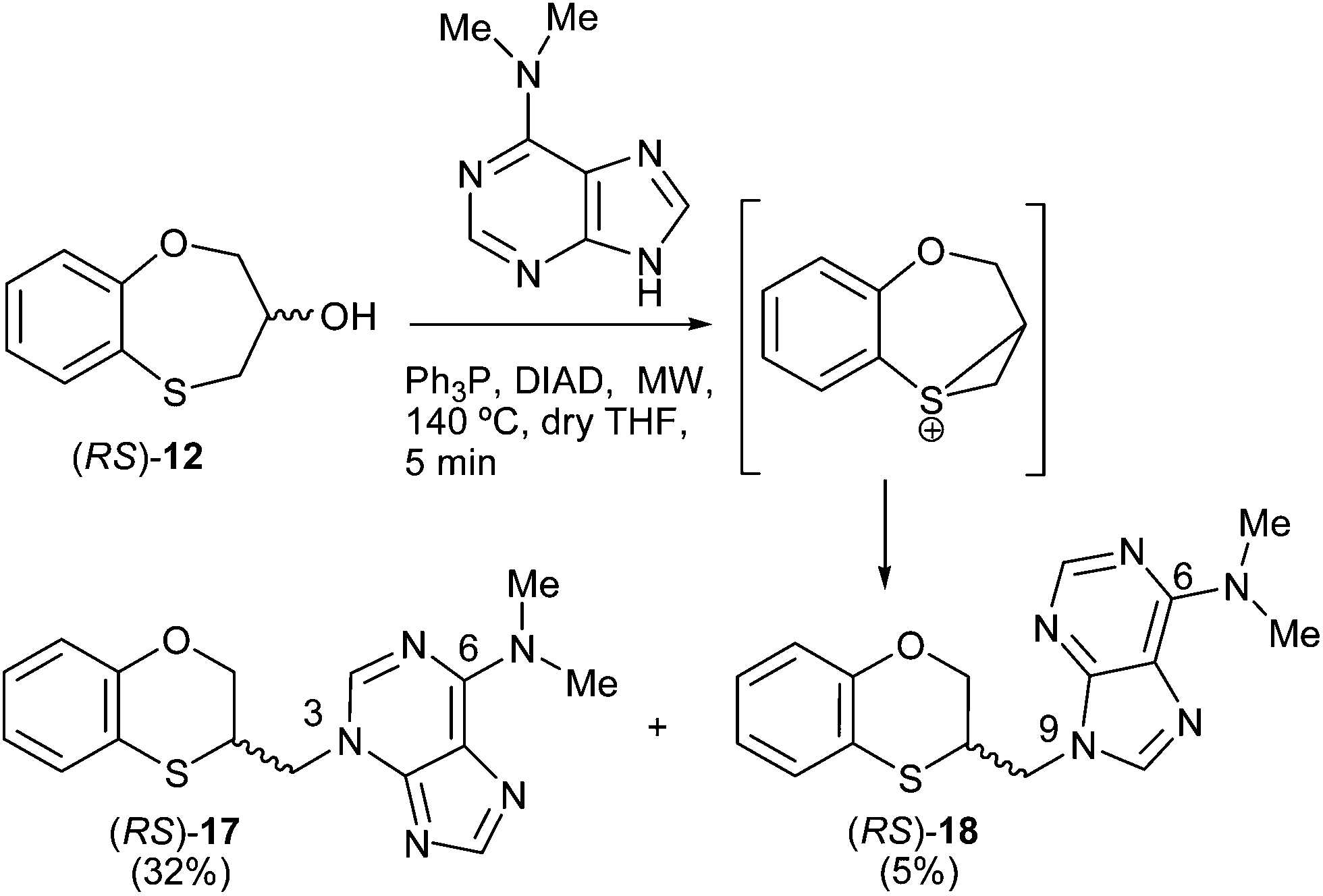 | ||
| Scheme 4 Preparation of the (RS)-(3-alkylated-N,N-dimethyl)adenine [(RS)-17] and (RS)-(9-alkylated-N,N-dimethyl)adenine [(RS)-18]. | ||
When (S)-12 (ee = 95.3%) is the starting reactant, the products obtained are (R)-17 (38%) and (R)-18 (5%), both of them with excellent optical purities (ee = 96.9% and ee = 94.2%, respectively) (Scheme 5).
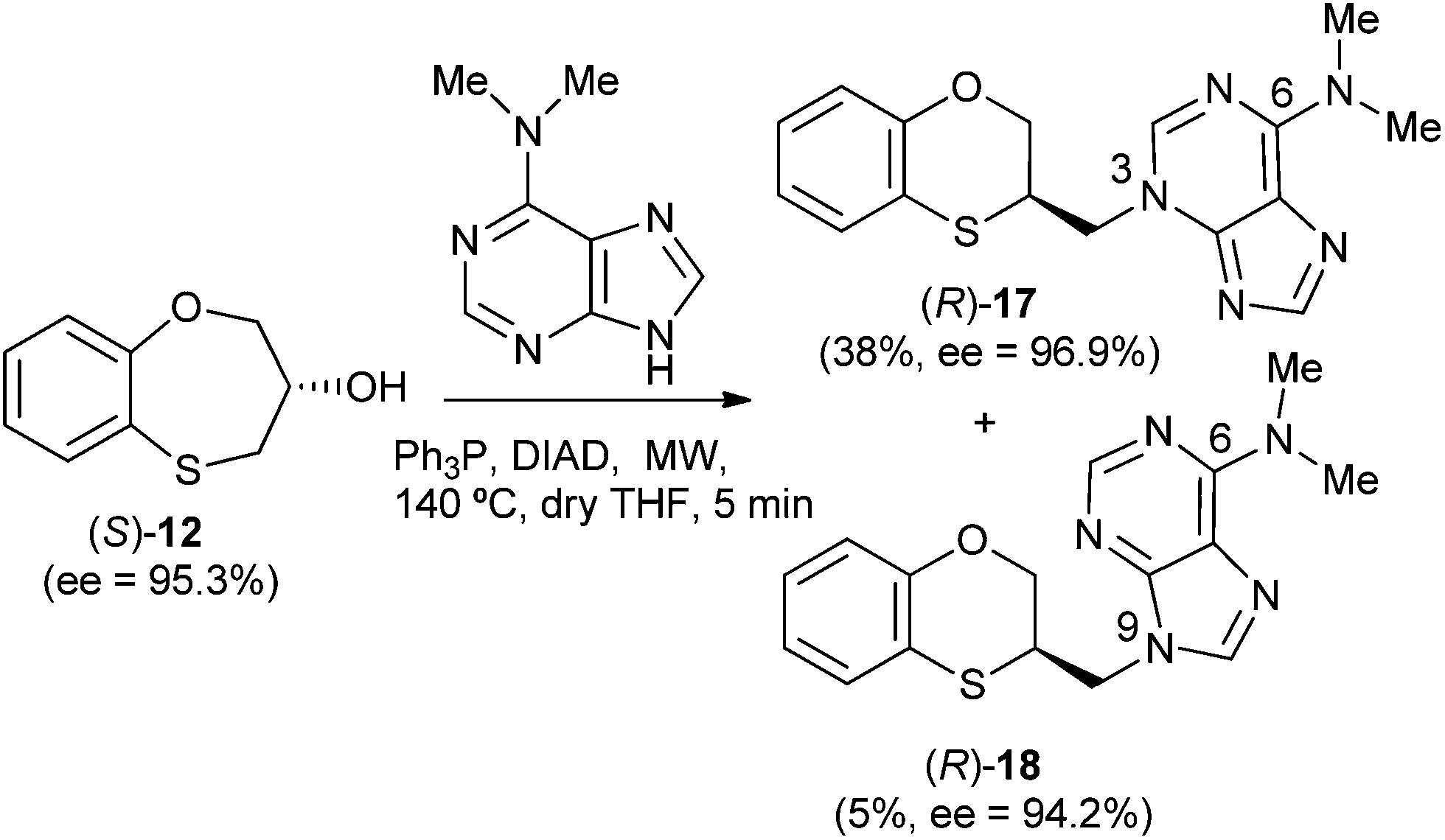 | ||
| Scheme 5 Reactivity between (S)-12 and N,N-dimethyladenine. | ||
When (R)-12 (ee = 74.7%) is the starting reactant, (S)-17 (34%, ee = 76%) and (S)-18 (5%, ee = 73.7%) are obtained (Scheme 6).
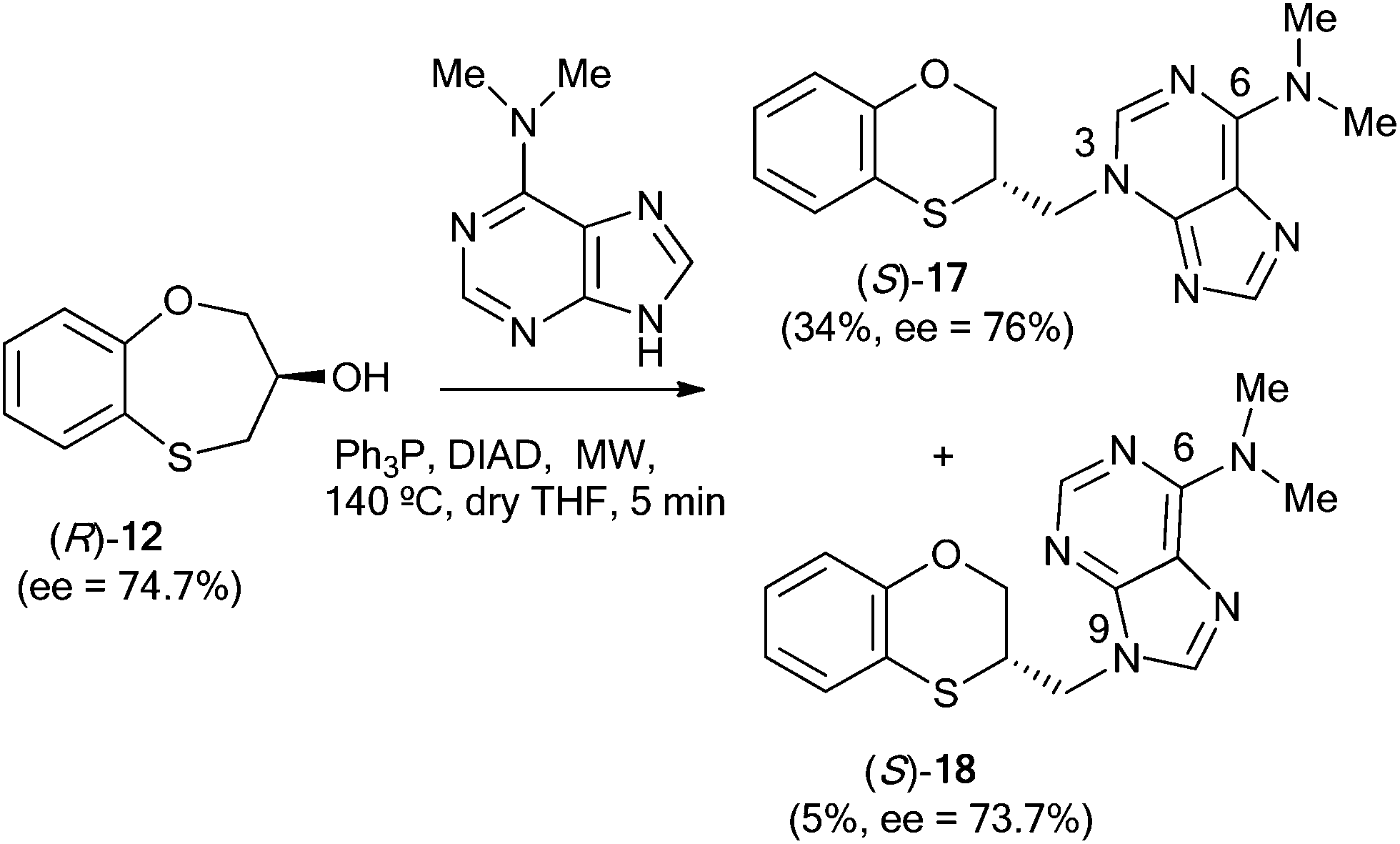 | ||
| Scheme 6 Reactivity between (R)-12 and N,N-dimethyladenine. | ||
The structure of 3-substituted-6-N,N-dimethyladenine is conditioned by the importance of the dipolar iminium form (20) to the possible resonance hybrid [19 ↔ 20 ↔ 21] (Scheme 7), as suggested by non-equivalent N-Me2 signals in the 1H NMR spectrum of 17 and is substantiated by its X-ray analysis. This is consistent with the electron-releasing nature of the dimethylamino group: σp = −0.83,18 or −0.82.19 Although the electronic influence of the NMe2 group on N-1 and N-3 of the purine nucleus is basically the same (its mesomeric electron-releasing effect is much more important than its inductive electron-withdrawing one), the lack of alkylation of N,N-dimethyladenine at its N-1 site could be attributed to the steric hindrance caused by the dimethylamino group. On the other hand, its steric influence also avoids the alkylation at the N-7 position of the adenine-derived compound.
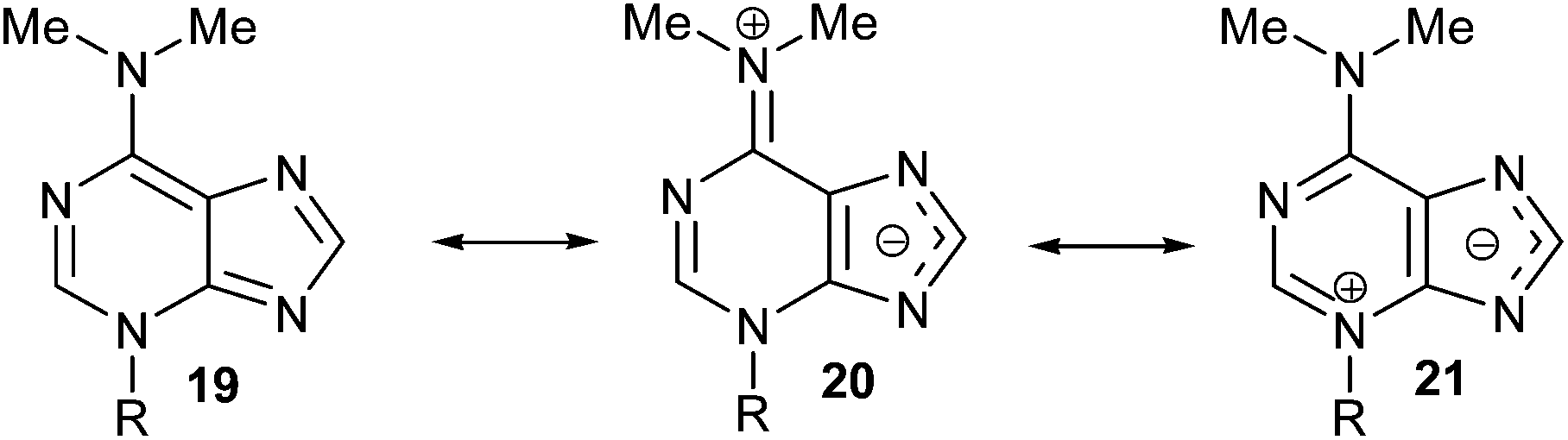 | ||
| Scheme 7 Resonance hybrid 20 can explain the alkylation at the N-3 atom of the purine moiety, as suggested by non-equivalent –NMe2 signals in the 1H NMR spectrum of (RS)-17. | ||
The structure of (RS)-17 has been determined by 1H, 13C NMR, HMBC (Fig. 2), HSQC and moreover by X-ray crystallography. HMBC gives 2-bond and 3-bond information, i.e., which of the 1H are 2-bonds or 3-bonds away from a particular 13C. HSQC gives 1-bond information, i.e., which 1H is attached to which 13C. The combination of 1H NMR and 1H-13C HSQC spectra is very powerful, and often allows the complete assignment of all protons in a molecule. In the HMBC experiment to three bonds, the following two correlations are important: (a) the first one between the exocyclic methylene group and the quaternary carbon C-4 of the purine (common carbon atom that is correlated through three bonds to CH-2 and CH-8) of 17; and the second one: (b) between the exocyclic methylene group and CH-2 (which is correlated to C-6) of the purine ring. This correlation proves unequivocally that the linkage between the six-membered moiety and the purine base takes place through N-3 in (RS)-17 (Fig. 2).
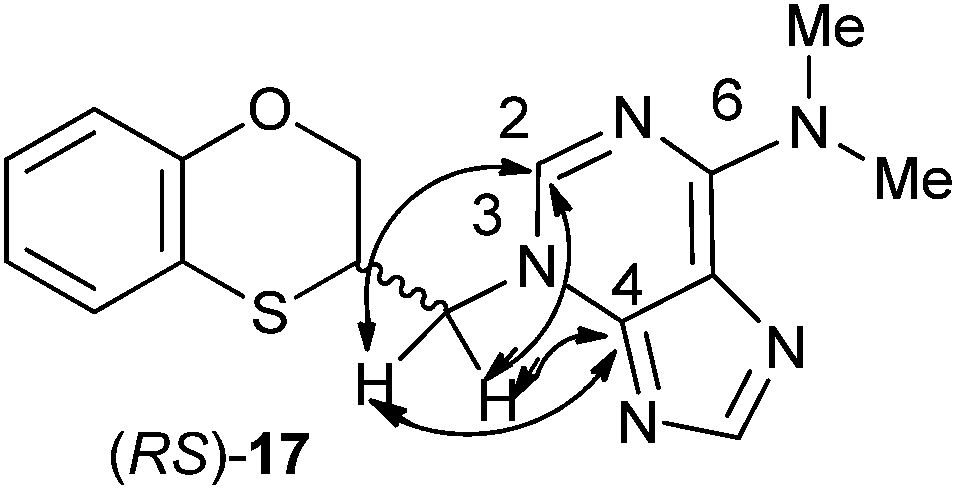 | ||
| Fig. 2 Representation of the HMBC interactions (with double-tipped arrows) observed between the exocyclic methylene group and the purine atoms, when the linkage is N-3 in compound (RS)-17. | ||
When the same reaction is carried out between (RS)-12, (R)-12, (S)-12 and N,N-dimethyladenine, the 3-substitution on N,N-dimethyladenine is still preferred, and is accompanied by the formation of the corresponding 9-substituted-N,N-dimethyladenine (RS)-18, (S)-18 and (R)-18. Miyaki and Shimizu20 found that, after chromatographic separation of the reaction products, the benzylation of N,N-dimethyladenine with PhCH2OH in THF gave the N-3 and N-9-regioisomers.
The structure of (RS)-18 has been determined by 1H, 13C NMR, HMBC and HSQC (Fig. 3). In the HMBC experiment to three bonds the following two correlations are important: (a) the first one between the exocyclic methylene group and the quaternary carbon C-4 of the purine (common carbon atom that is correlated through three bonds to CH-2 and CH-8) of (RS)-18 and the second one: (b) between the exocyclic methylene group and CH-8 (which is the CH group that is not correlated with C-6) of the purine ring. This correlation proves unequivocally that the linkage between the six-membered moiety and the purine base takes place through N-9 in (RS)-18 (Fig. 3).
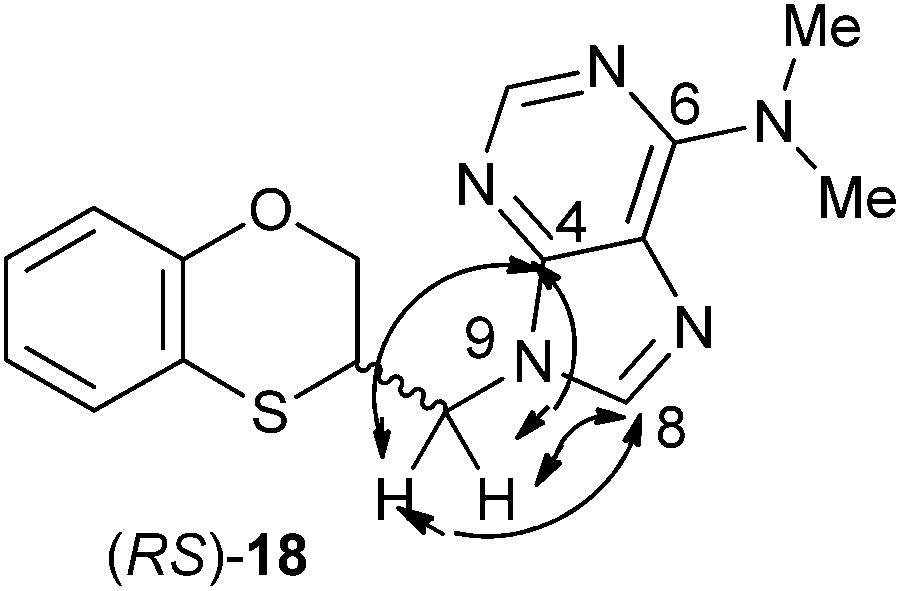 | ||
| Fig. 3 Representation of the HMBC interactions (with double-tipped arrows) observed between the exocyclic methylene group and the purine atoms, when the linkage is N-9 in compound (RS)-18. | ||
Reactivity between racemic and enantiomers of 4-dihydro-2H-1,5-benzoxathiepin-3-ol and 2-chloro-N-methyladenine
The Mitsunobu coupling of allylic and benzylic alcohol with adenine and 2-chloroadenine has been reported previously with good N-9 selectivity.21–23 However, poor to modest yield (20–60%) and limited substrate scope were observed,24–26 which significantly limits the application of this method. Currently, there is no detailed study regarding the Mitsunobu coupling between 2-chloroadenine and a secondary alcohol through neighbouring-group participation.The methylamino moiety is an electron-releasing group with a σp similar to that of the dimethylamino moiety {σp (MeHN) = −0.76;21 σp (Me2N) = −0.83,18 or 0.82 (ref. 19)} and then favours the nucleophilicity of the N-3 atom of purine. However the presence of an electron-withdrawing atom such as chlorine {σI (Cl) = 0.47 (ref. 27)} neutralizes the effect of the methylamino group and consequently the alkylation site moves to the N-9 atom of 2-chloro-N-methyladenine, i.e., going from 17 and 18 to 22, there is a shift of σ-electron-density from the six- to the five-membered ring of purine. Moreover, no exocyclic N-alkylation takes place due to its relatively weak nucleophilic character.
The structure of (RS)-22 has been determined by 1H, 13C NMR, HMBC and by X-ray in (R)-22 (Schemes 8–10). The HMBC experiment to three bonds proves unequivocally that the linkage between the six-membered moiety and the purine base does not take place through N-3 because the exocyclic methylene group is correlated with CH-8 of the purine, and accordingly the linkage could be through N-9 or N-7 (Fig. 4). It was finally determined by (R)-22 X-ray crystal structure because in the HMBC experiment it was impossible to differentiate between C-4 and C-5 of the purine.
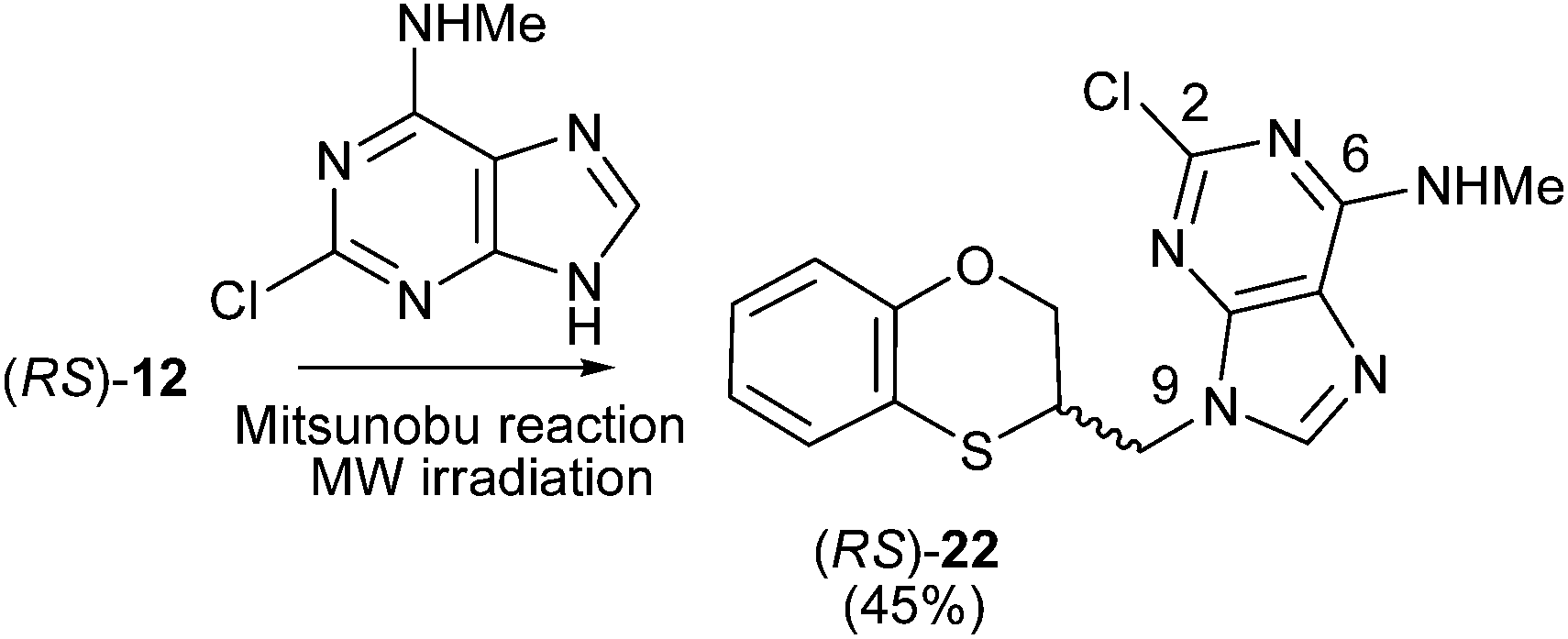 | ||
| Scheme 8 Preparation of (RS)-(N-9-alkylated-2-chloro-N-methyl)adenine [(RS)-22]. | ||
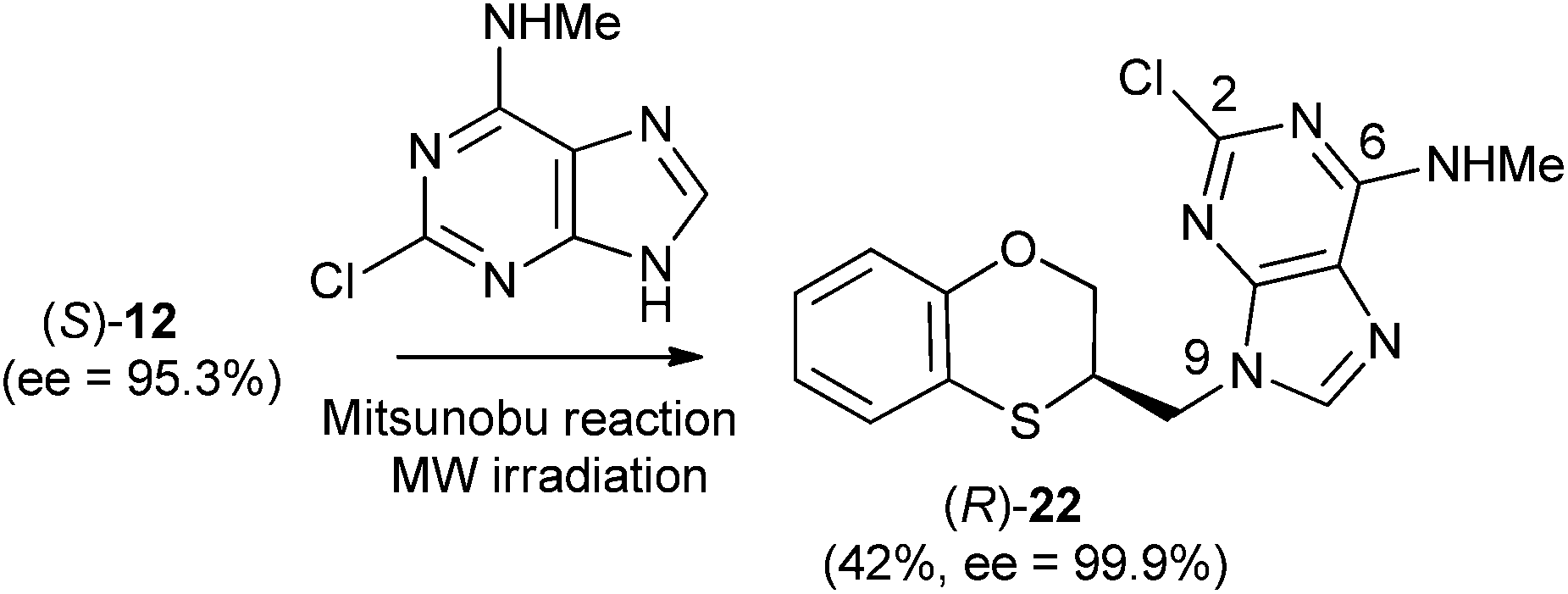 | ||
| Scheme 9 Preparation of (R)-(N-9-alkylated-2-chloro-N-methyl)adenine [(R)-22]. | ||
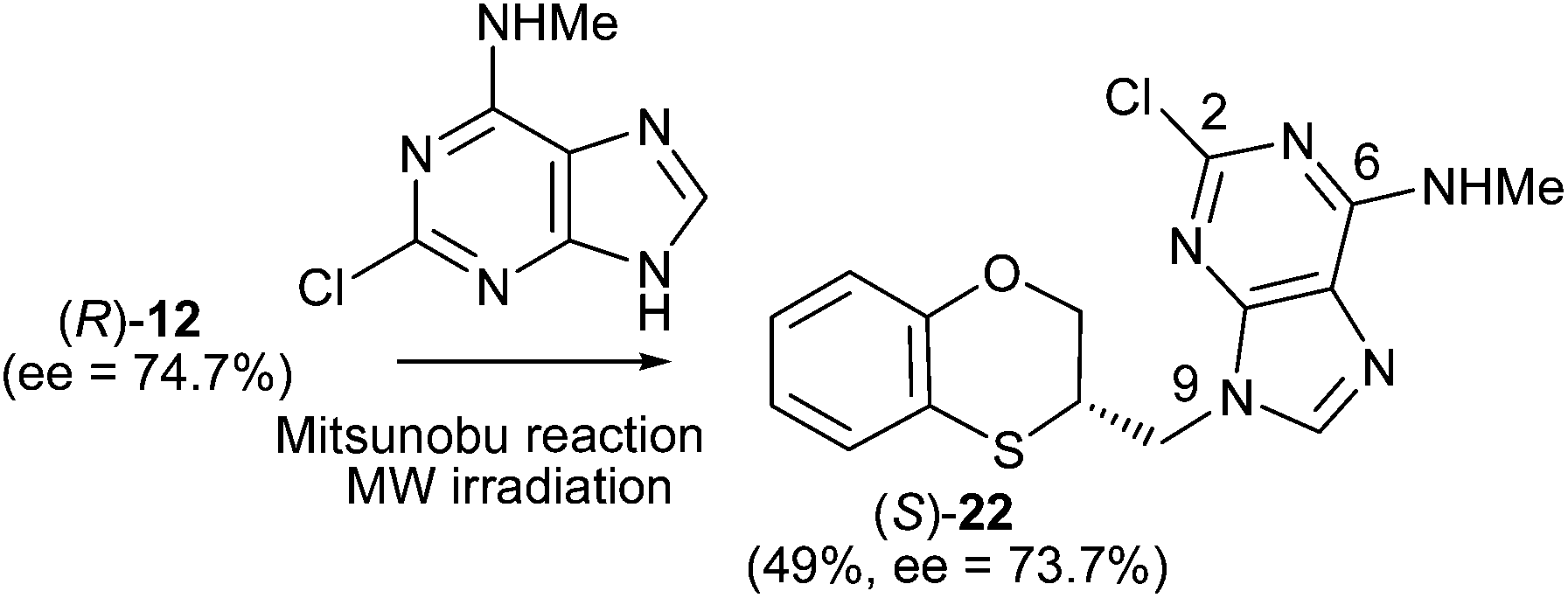 | ||
| Scheme 10 Preparation of (S)-(N-9-alkylated-2-chloro-N-methyl)adenine [(S)-22]. | ||
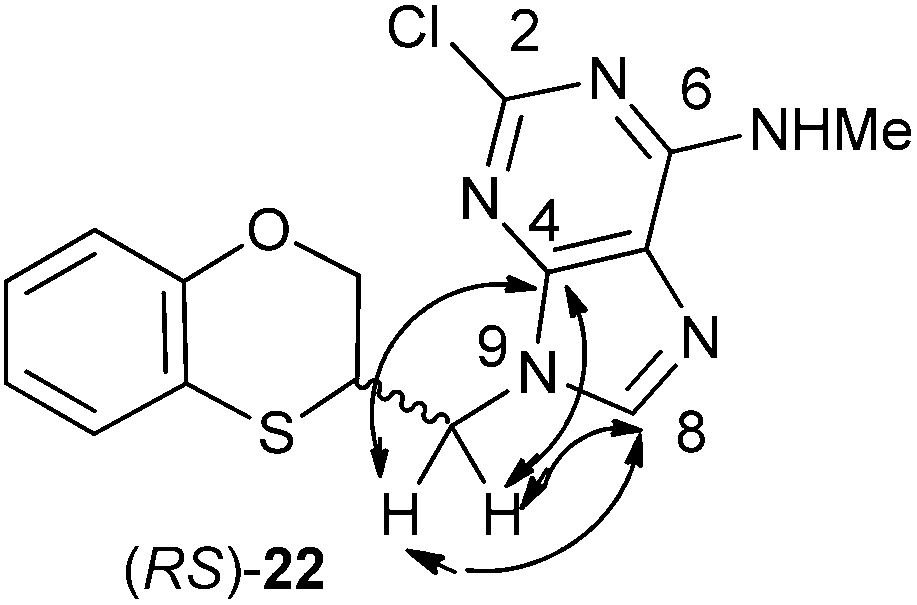 | ||
| Fig. 4 Representation of the HMBC interactions (with double-tipped arrows) observed between the exocyclic methylene group and the purine atoms of (RS)-22 when the linkage is through N-9. | ||
The stereochemistry of (S)-22 shows the complete inversion of the stereogenic alcohol center. This result is consistent with a SN2 mechanism and strongly enhances the application of the reported method as a general approach for the stereospecific synthesis of alkylated purines.
Reactivity between racemic and enantiomers of 4-dihydro-2H-1,5-benzoxathiepin-3-ol and N-methyladenine
Reactions between (RS)-12, (R)-12 or (S)-12 and N-methyladenine produce the same regioisomers (N-9 and N-3) and the sum of the global yields of the final products are in the same range [90% from (RS)-12, 89% from (S)-12 and 90% from (R)-12] (Schemes 11–13). Reactions between (RS)-12, (R)-12 and (S)-12 and 6-(N-methyl)adenine produce the regioisomers (RS)-23 (N-3: 45%) and (RS)-24 (N-9: 45%), (S)-23 (N-3: 48%) and (S)-24 (N-9: 42%), (R)-23 (N-3: 47%) and (R)-24 (N-9: 42%), respectively. This is consistent with the electron-releasing nature of the methylamino group: σp = −0.76.19 In the case of the NHMe group compared to the NMe2 one, the lesser electron-releasing power of the former in relation to the latter explains the approximately equal amount of both N-3 (23) and N-9 (24) regioisomers in the reaction between 12 and N-methyladenine, whilst in the case of N,N-dimethyladenine the major product is the N-3 regioisomer (RS)-, (R)-, and (S)-17, being the ratio N-9/N-3 ≈ 7/1.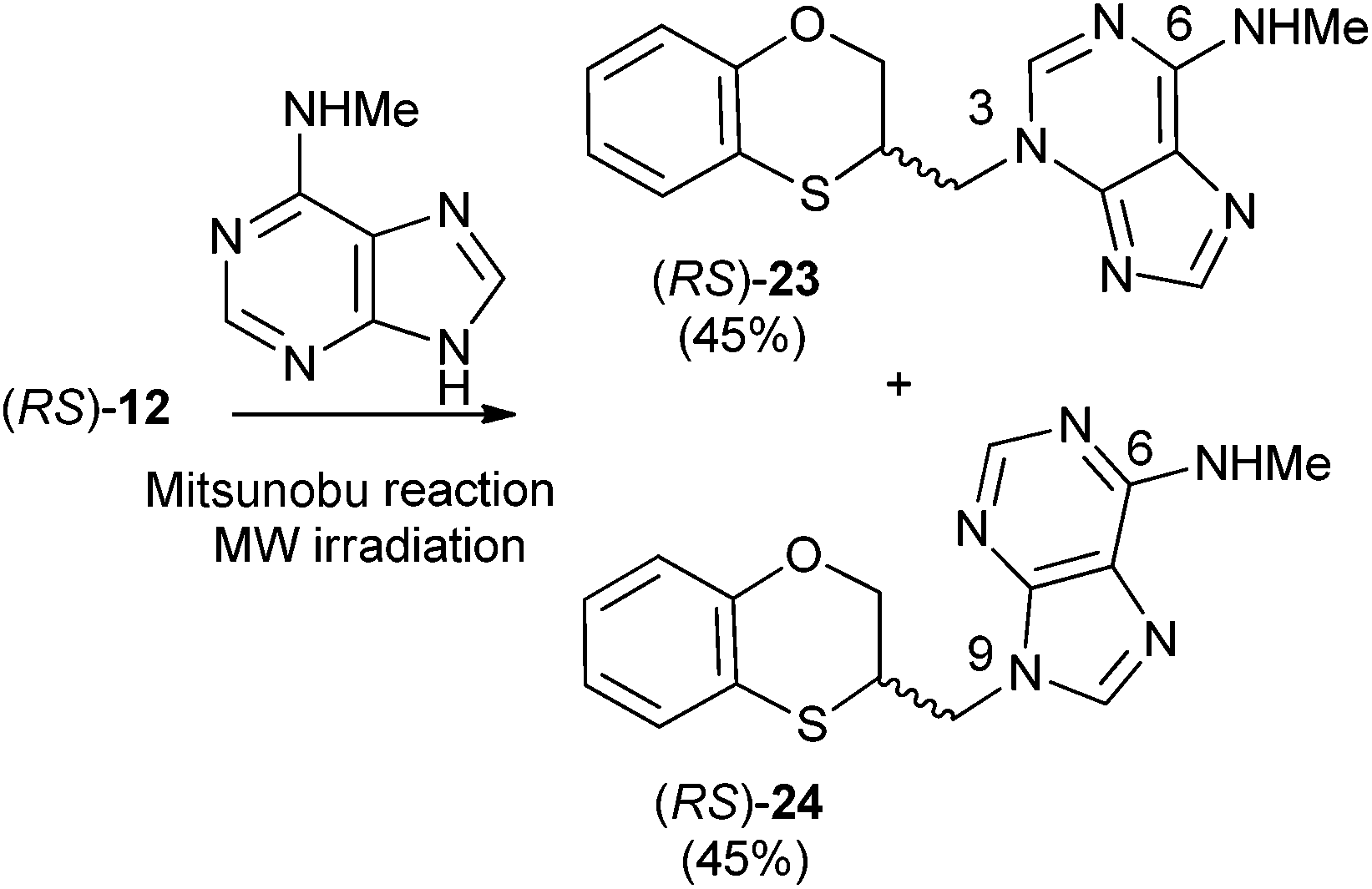 | ||
| Scheme 11 Preparation of (RS)-(N-3-alkylated-N-methyl)adenine [(RS)-23] and (RS)-(N-9-alkylated-N-methyl)adenine [(RS)-24]. | ||
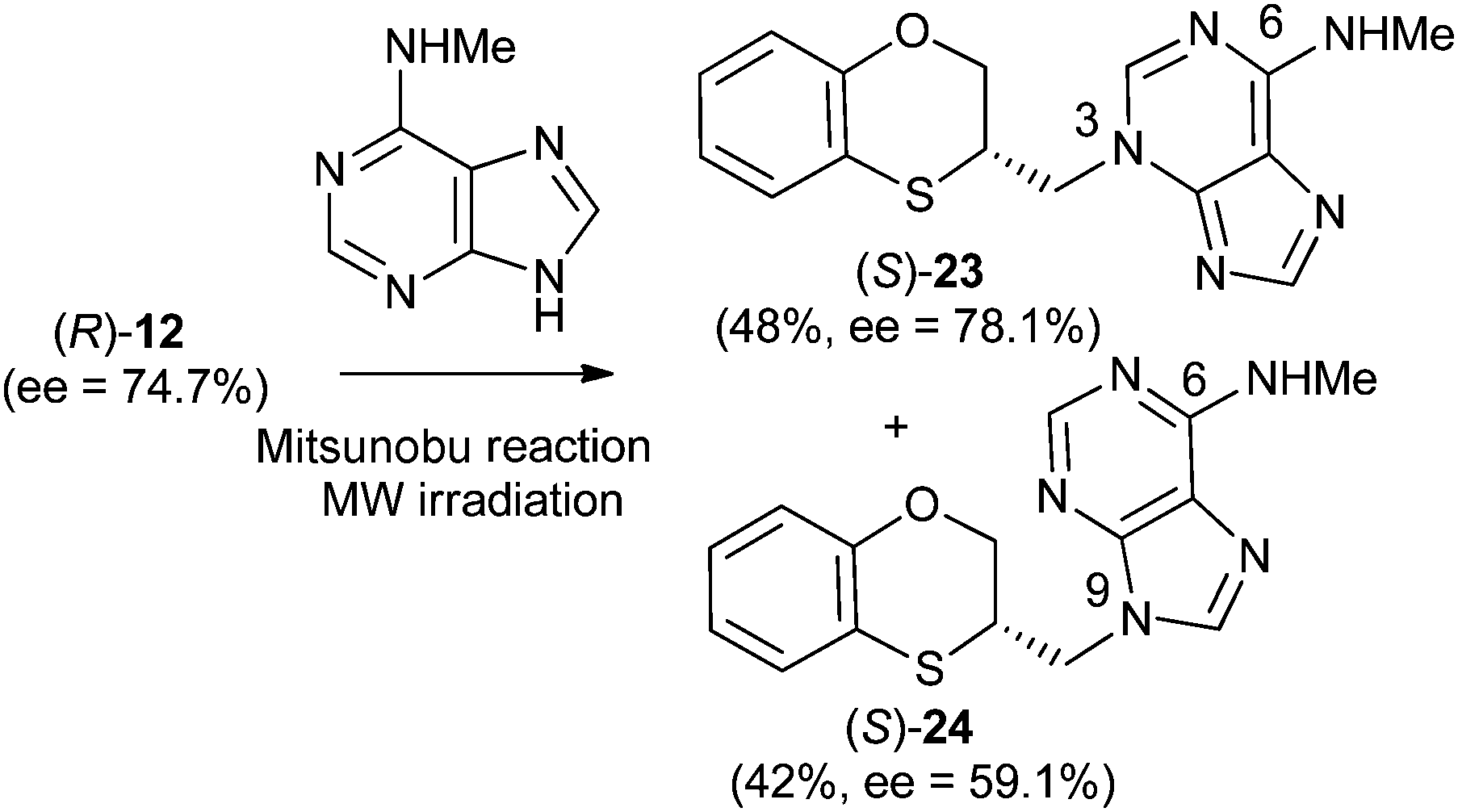 | ||
| Scheme 12 Preparation of (S)-(N-3-alkylated-N-methyl)adenine [(S)-23] and (S)-(N-9-alkylated-N-methyl)adenine [(S)-24]. | ||
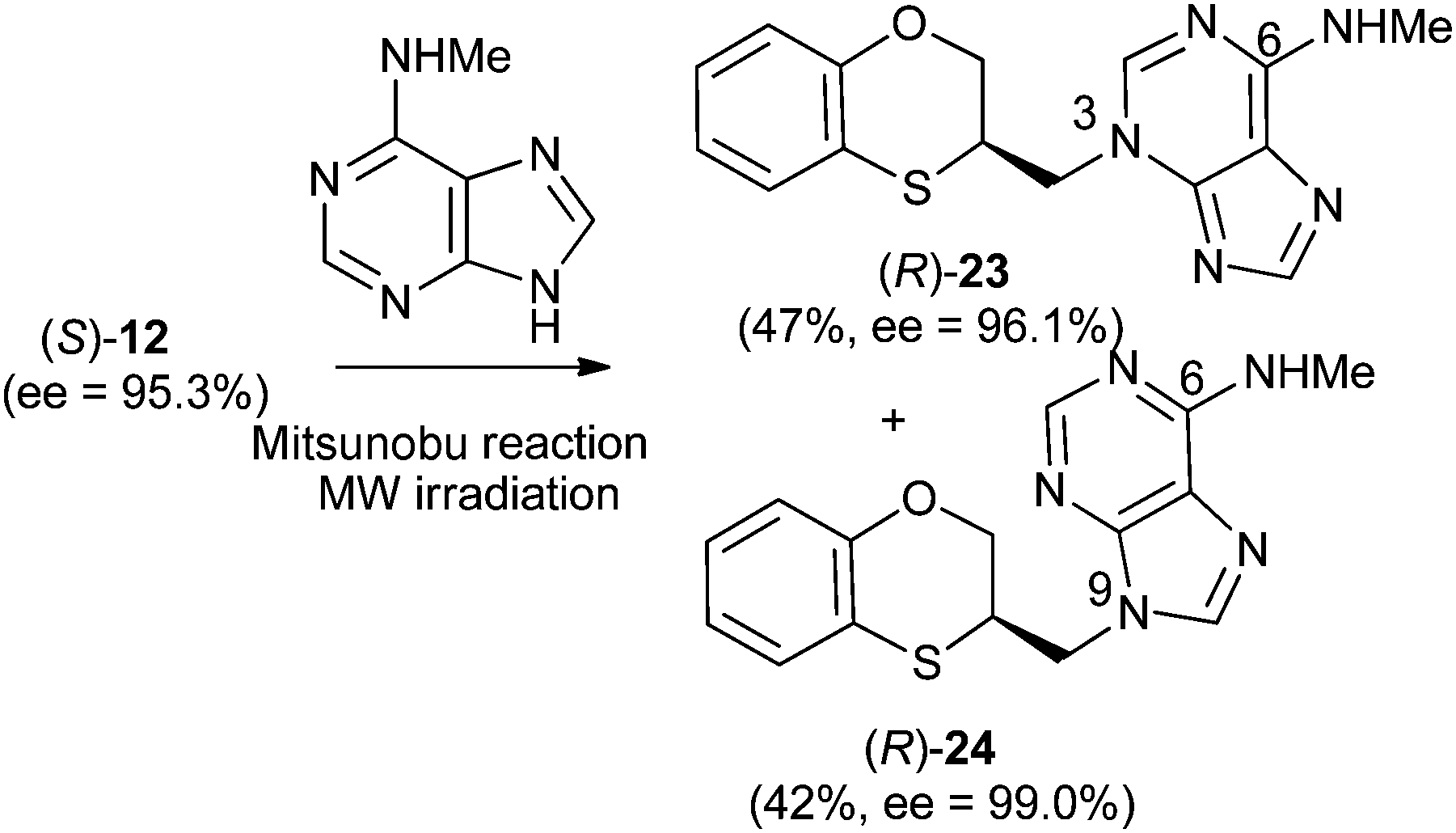 | ||
| Scheme 13 Preparation of (R)-(N-3-alkylated-N-methyl)adenine [(R)-23] and (R)-N-9-alkylated-N-methyl)adenine [(R)-24]. | ||
In the HMBC experiment to three bonds, the two following correlations are important: (a) the first one between the exocyclic methylene group and the quaternary carbon C-4 of the purine (common carbon atom which is correlated to CH-2 and CH-8) of (RS)-23; and the second one: (b) between the exocyclic methylene group and CH-2 (which is the group that is not correlated with C-6) of the purine ring. This correlation proves unequivocally that the linkage between the six-membered moiety and the purine base takes place through N-3 in (RS)-23 (Fig. 5).
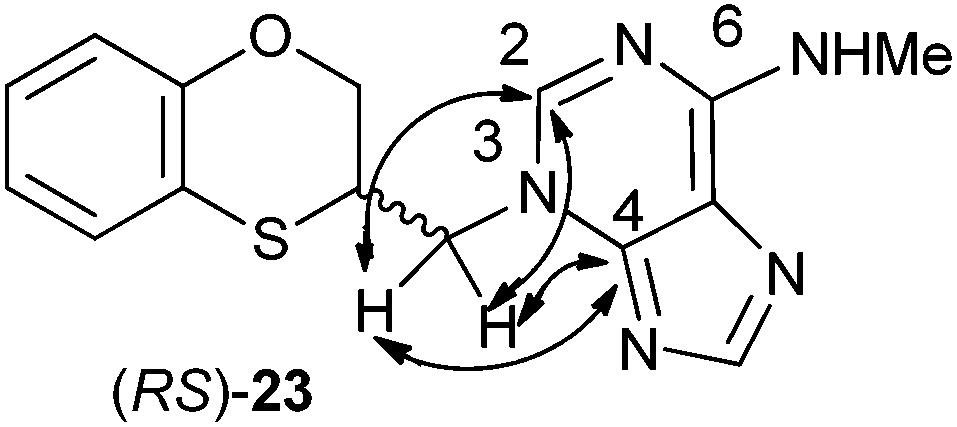 | ||
| Fig. 5 Representation of the HMBC interactions (with double-tipped arrows) observed between the exocyclic methylene group and the purine atoms, when the linkage is through N-3 in compound (RS)-23. | ||
The structure of (RS)-24 has been determined by 1H, 13C NMR, HMBC and HSQC. In the HMBC experiment to three bonds the following two correlations are outstanding: (a) the first one between the exocyclic methylene group and the quaternary carbon C-4 of the purine (common carbon atom that is correlated through three bonds to CH-2 and CH-8) of (RS)-24; and the second one: (b) between the exocyclic methylene group and CH-8 (which is the CH group that is not correlated with C-6) of the purine ring. This correlation proves unequivocally that the linkage between the six-membered moiety and the purine base takes place through N-9 in (RS)-24 (Fig. 6).
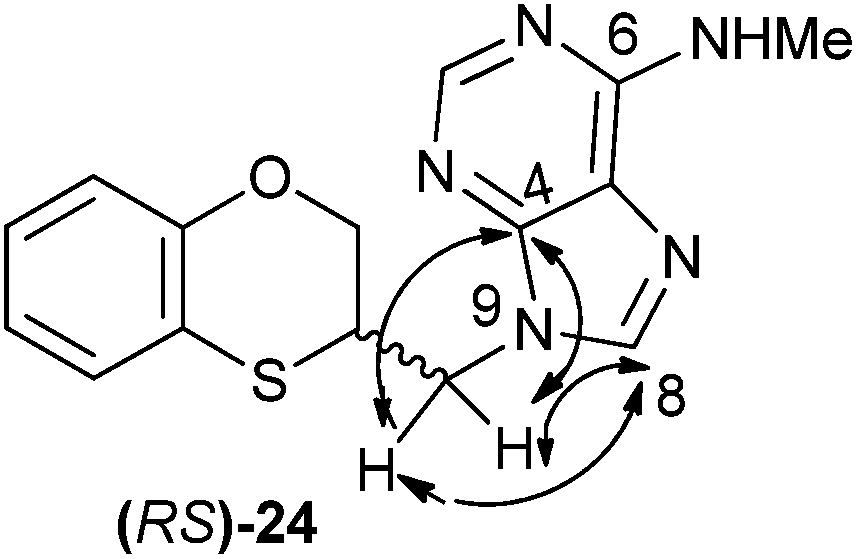 | ||
| Fig. 6 Representation of the HMBC interactions (with double-tipped arrows) observed between the exocyclic methylene group and the purine atoms, when the linkage is through N-9 in compound (RS)-24. | ||
Neither 2,6-diaminopurine nor 6,8-dichloro-2-methylthiopurine, as possible purine bases in the Mitsunobu coupling, produced the desired coupling products with the secondary racemic and homochiral alcohols 12. These results may be due to the poor solubility of the bases.
NMR data for adenine-derived compounds
The 1H NMR (CDCl3) CH-2 signal of adenines in N-9 isomer (RS)-18 is shifted downfield in relation to the corresponding signal of the N-3 isomer (RS)-17, whereas the CH-8 signal of (RS)-18 is shifted upfield compared with the corresponding signal of N-3 [(RS)-17]. In summary, the signals of both proton atoms are interchanged in both regioisomers. Compounds (RS)-23 and -24 cannot be compared because the 1H NMR spectra of both (RS)-23 and -24 were registered in different deuterated solvents (CDCl3 and DMSO-d6, respectively).Chemical shifts (CDCl3) of the carbon atom for the CH-2 group follows the same tendency in N-3 [(RS)-17 and -23] and N-9 [(RS)-18 and -24] isomers. These signals are shifted downfield in N-3 isomers [(RS)-17 and (RS)-23, δ 141.9–145.1 ppm] compared with the corresponding signals in N-9 isomers [(RS)-18 and -24, δ 152.8–152.4 ppm].
Chemical shift (CDCl3) of the carbon atom for CH-8 is shifted up-field in N-9 isomers [(RS)-18 and (RS)-24, δ 152.7–148.2 ppm] compared with the corresponding signals in N-3 isomers [(RS)-17 and -23, δ 139.5–140.7 ppm].
Experimental
General methods
Melting points were taken in open capillaries and are uncorrected. Flash chromatography was performed on silica gel 60 with a particle size of 0.040–0.063 mm (230–400 mesh ASTM). Nuclear magnetic resonance spectra have been recorded on a 600 MHz 1H and 150 MHz 13C NMR, 500 MHz 1H and 125 MHz 13C NMR, 400 MHz 1H and 100 MHz 13C NMR or 300 MHz 1H and 75 MHz 13C NMR spectrometers at ambient temperature. Chemical shifts (δ) are quoted in parts per million (ppm) and are referenced to the residual solvent peak. Signals are designated as follows: s, singlet; bs, broad singlet, d, doublet; dd, doublet of doublets; ddd, double doublet of doublets; t, triplet; m, multiplet. The HMBC spectra were measured using a pulse sequence optimized for 10 Hz (inter-pulse delay for the evolution of long-range couplings: 50 ms) and a 5/3/4 gradient combination. In this way, direct responses (J couplings) were not completely removed. High-resolution Nano-Assisted Laser Desorption/Ionization (NALDI-TOF) or Electrospray Ionization (ESITOF) mass spectra were carried out. Small scale microwave-assisted synthesis was carried out in a single-mode microwave instrument producing controlled irradiation at 2.450 GHz and sealed reaction vessels were used. Reaction time refers to hold time from 130 °C to 160 °C, not to total irradiation time. The temperature was measured with an IR sensor outside the reaction vessel. Anhydrous DMF was used as received. Anhydrous conditions were performed under argon. X-ray data were collected on a diffractometer equipped with graphite a monochromated Mo Kα radiation. The structure was solved by direct methods shelxs97 and refined by full-matrix least-squares against F2 using shelxl97 software.Preparation of (S)-3-[(2-hydroxyphenyl)thio]-1,2-epoxypropane [(S)-14]
A mixture of 2-mercaptophenol (200 mg, 1.59 mmol) and 60% pyridine (130 mg, 1.59 mmol) in anhydrous DMF (2.5 mL) under argon, was stirred at r.t. for 5 min, (R)-epichlorohydrin (170 mg, 1.83 mmol) was added and the resulting mixture was irradiated under microwaves conditions at 140 °C for an additional 10 min. The mixture was cooled, and the pH was fixed to 2–3 with 1 N HCl. The DMF was removed in vacuo, and the residue was extracted (H2O–CH2Cl2). Organic layers were combined and dried (Na2SO4). After filtration the solvent was removed in vacuo and the crude was purified by flash column chromatography using an 8/2 hexane–EtOAc mixture. (S)-14 (260 mg, 90%) was obtained as a brownish syrup. ee = 99.9%, [α]25D −46.6 (c 1.0 in CH2Cl2); HPLC analysis: hexane–2-propanol = 90/10, flow rate 1.0 mL min−1, λ = 250 nm, tR (S)-14 = 11.346 min.Preparation of (S)-3,4-dihydro-2H-1,5-benzoxathiepin-3-ol [(S)-12] and (R)-2,3-dihydro-1,4-benzoxathiin-2-methanol [(R)-13]
Compounds (S)-12 (62 mg, 62%) and (R)-13 (24 mg, 24%) were synthesized according to our previously reported procedure for the racemic compounds,5,14 but using (R)-epichlorohydrin instead. (S)-12: ee = 95.3%, [α]25D −36.7 (c 1.0 in CH2Cl2); HPLC analysis: hexane–CH2Cl2 = 65/35, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-12 = 10.896 min, tR (S)-12 = 12.079 min [Fig. (S3)†].(R)-13: ee = 98.5%, [α]25D + 38.1 (c 1.0 in CH2Cl2); HPLC analysis: hexane–CH2Cl2 = 65/35, flow rate 1.0 mL min−1, λ = 250 nm, tR (S)-13 = 14.682 min, tR (R)-13 = 15.890 min.
Preparation of (R)-12 and (S)-13
The procedure was exactly the same as described to obtain (S)-12 and (R)-13, but compound (R)-14 was not isolated, NaOH (63 mg, 1.59 mmol) and more DMF (10 mL) were added (instead of water) and the resulting solution was heated at 100 °C for 24 h. After removing DMF and subsequent flash chromatography, (R)-12 (179 mg, 62%) and (S)-13 (69 mg, 24%) were obtained.(R)-12: ee = 74.7%, [α]25D + 28.3 (c 1.0 in CH2Cl2); HPLC analysis: hexane–CH2Cl2 = 65/35, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-12 = 9.854 min, tR (S)-12 = 11.114 min.
(S)-13: ee = 77.3%, [α]25D −33.2 (c 1.0 in CH2Cl2); HPLC analysis: hexane–CH2Cl2 = 65/35, flow rate 1.0 mL min−1, λ = 250 nm, tR (S)-13 = 14.899, tR (R)-13 = 16.249 min.
General procedure for the microwave-assisted synthesis of racemic and homochiral compounds 17, 18, 22, 23 and 24
DIPAD (2 equiv.) was slowly added to a suspension of the benzo-fused six-membered ring 12 (1 equiv.), the purine base (1 equiv.) and triphenylphosphine (1.1 equiv.) in dry THF (5 mL mmol−1) in a microwave vial, cooled in an ice-bath. The vial was sealed and microwave irradiated at a temperature of 140 °C for 10 min. Compounds 17, 18, 22, 23 and 24 were purified by flash column chromatography on silica gel using a mixture of hexane–EtOAc (5/1 → 0/1) and then CH2Cl2–MeOH (10/0.1 → 7/3). The solvent was evaporated in vacuo and the products were obtained pure.(RS)-N,N-Dimethyl-3-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-3H-adenine [(RS)-17]: white solid (57 mg, 32%), mp 152–154 °C. 1H NMR: δH (500 MHz, CDCl3) 3.35 (3H, br s, CH3), 3.92 (3H, br s, CH3), 4.18 (1H, m, CH-3), 4.27 (1H, d, Jgem = 12.0 Hz, J1,2 = 1.7 Hz, CH-2), 4.37 (1H, dd, Jgem = 12.0 Hz, J1,2 = 2.9 Hz, CH-2), 4.42 (1H, dd, Jgem = 13.7 Hz, J1,2 = 8.4 Hz, 1H-exocyclic), 4.65 (1H, dd, Jgem = 13.7 Hz, J1,2 = 6.9 Hz, 1H-exocyclic), 6.89 (2H, m, CH-aromatics), 7.03 (2H, m, CH-aromatics), 7.87 (1H, s, H2-purine), 7.96 (1H, s, H8-purine). 13C NMR: δC (125 MHz, CDCl3) 36.99 (CH-3), 38.23 (CH3), 39.95 (CH3), 51.77 (CH2-exocyclic), 66.14 (CH2-2), 115.92 (C-4a), 122.02 (C-5-purine), 118.91 (CH-aromatics), 122.74, 126.38, 128.15, 141.92 (CH-2-purine), 150.11 (C-4-purine), 151.27 (C-8a), 152.74 (CH-8-purine), 153.85 (C-6-purine). HR LSIMS m/z calcd for C16H17N5OS [M + H]+ 328.1232, found 328.1238.
(R)-N,N-Dimethyl-3-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-3H-adenine [(R)-17] (68 mg, 38%): ee = 96.9%, [α]25D −135.49 (c 1.0 in CH2Cl2); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-17 = 17.84 min, tR (S)-17 = 23.430 min.
(S)-N,N-Dimethyl-3-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-3H-adenine [(S)-17] (61 mg, 34%): ee = 76%, [α]25D + 104.74 (c 1.0 in CH2Cl2); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-17 = 18.242 min, tR (S)-17 = 23.055 min.
(RS)-N,N-Dimethyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(RS)-18]: white solid (9 mg, 5%), mp 170–172 °C. 1H NMR: δH (500 MHz, CDCl3) 3.52 (6H, br s, N(CH3)2), 3.89 (1H, m, CH-3), 4.25 (2H, m, CH2-2), 4.41 (1H, dd, Jgem = 14.2 Hz, J1,2 = 7.7 Hz, 1H-exocyclic), 4.50 (1H, dd, Jgem = 14.2 Hz, J1,2 = 7.7 Hz, 1H-exocyclic), 6.89 (2H, m, CH-aromatics), 7.02 (2H, m, CH-aromatics), 7.71 (1H, s, H8-purine), 8.33 (1H, s, H2-purine). 13C NMR: δC (125 MHz, CDCl3) 29.93 (CH3), 37.72 (CH-3), 45.76 (CH2-exocyclic), 65.91 (CH2-2), 116.25 (C-4a), 118.82 (CH-aromatics), 120.54 (C-5-purine), 122.63, 126.17, 127.96, 139.15 (CH-8-purine), 150.65 (C-4-purine), 151.45 (C-8a), 152.84 (CH-2-purine), 155.25 (C-6-purine). HR LSIMS m/z calcd for C16H17N5OS [M + H]+ 328.1232, found 328.1233.
(R)-N,N-Dimethyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(R)-18] (9 mg, 5%): ee = 94.2%, [α]25D + 67.1 (c 0.34 in MeOH); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-18 = 20.053 min, tR (S)-18 = 25.755 min.
(S)-N,N-Dimethyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(S)-18] (9 mg, 5%): ee = 73.7%, [α]25D −53.3 (c 0.34 in MeOH); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-18 = 20.050 min, tR (S)-18 = 25.764 min.
(RS)-2-Chloro-N-methyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(RS)-22]: white solid (86 mg, 45%), mp 117–119 °C. 1H NMR: δH (500 MHz, CDCl3) 3.11 (3H, m, CH3), 3.82 (1H, m, CH-3), 4.23 (1H, ddd, Jgem = 12.0 Hz, J1,2 = 3.0 Hz J1,3 = 1.9 Hz, CH-3), 4.25 (2H, m, CH2-2), 4.37 (1H, dd, Jgem = 14.2 Hz, J1,2 = 8.0 Hz, CH2 exocyclic), 4.47 (1H, dd, Jgem = 14.2 Hz, J1,2 = 7.5 Hz, CH2 exocyclic), 6.88 (2H, m, CH-aromatics), 7.02 (2H, m, CH-aromatics), 7.73 (1H, s, H8-purine). 13C NMR: δC (126 MHz, CDCl3) 27.75 (CH3), 37.53 (CH-3), 45.79 (CH2-exocyclic), 65.70 (CH2-2), 115.72 (C-4a), 118.21 (C-5-purine), 118.74 (CH-aromatics), 122.64, 126.20, 127.84, 140.45 (CH-8-purine), 149.63 (C-4-purine), 151.19 (C-8a), 155.86 (C-6-purine). HR LSIMS m/z calcd for C16H14N5ClOS [M + H]+ 348.0686, found 348.0687.
(R)-2-Chloro-N-methyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(R)-22] (85 mg, 42%): ee = 99.9%, [α]25D + 33.59 (c 0.65 in DMSO); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-22 = 13.353 min.
(S)-2-Chloro-N-methyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(S)-22] (100 mg, 49%): ee = 73.7%, [α]25D −22.8 (c 0.65 in DMSO); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-22 = 13.352 min, tR (S)-22 = 14.560 min.
(RS)-N-Methyl-3-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-3H-adenine [(RS)-23]: white solid (77 mg, 45%), mp 199–201 °C. 1H NMR: δH (500 MHz, CDCl3) 3.24 (3H, m, CH3), 4.08 (1H, m, H-3), 4.31 (1H, dd, Jgem = 12.1 Hz, J1,2 = 1.5 Hz, CH2-2), 4.40 (1H, dd, Jgem = 12.11 Hz, J1,2 = 2.8 Hz, CH2-2), 4.49 (1H, dd, Jgem = 13.7 Hz, J1,2 = 8.5 Hz, CH2-exocyclic), 4.72 (1H, dd, Jgem = 13.8 Hz, J1,2 = 6.7 Hz, CH2-exocyclic), 6.90 (2H, m), 7.04 (2H, m), 8.00 (1H, s, H8-purine), 8.07 (1H, s, H2-purine). 13C NMR: δC (126 MHz, CDCl3) 28.32 (CH3), 37.11 (CH-3), 51.81 (CH2-exocyclic), 65.90 (CH2-2), 115.12 (C4a), 117.19 (C-5-purine), 118.96 (CH-aromatics), 122.91, 126.59, 128.10, 145.14 (CH-2-purine), 147.47 (C-4-purine), 148.24 (CH-8-purine), 151.20 (C-8a), 153.86 (C-6-purine). HR (LSIMS) m/z calcd for C15H16N5OS [M + H]+ 314.1076, found 314.1076.
(R)-N-Methyl-3-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-3H-adenine [(R)-23] (80 mg, 47%): ee = 96.1%, [α]25D −61.81 (c 1.0 in MeOH); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-23 = 14.343 min, tR (S)-23 = 17.190 min.
(S)-N-Methyl-3-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-3H-adenine [(S)-23] (83 mg, 48%): 78.1 ee, [α]25D + 59.43 (c 1.0 in MeOH); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-23 = 15.004 min, tR (S)-23 = 17.299 min.
(RS)-N-Methyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(RS)-24]: white solid (77 mg, 45%), mp 168–170 °C. 1H NMR: δH (600 MHz, DMSO-d6) 2.95 (3H, s, CH3), 4.12 (1H, m, H-3), 4.28 (2H, m, CH2-2), 4.40 (1H, dd, Jgem = 14.1 Hz, J1,2 = 8.2 Hz, CH2-exocyclic), 4.56 (1H, dd, Jgem = 14.1 Hz, J1,2 = 6.7 Hz, CH2-exocyclic), 6.90 (2H, m), 7.06 (2H, m), 8.13 (1H, s, H8-purine), 8.23 (1H, s, H2-purine). 13C NMR: δC (150 MHz, DMSO d6) 26.96 (CH3), 37.82 (CH-3), 44.65 (CH2-exocyclic), 65.95 (CH2-2), 116.45 (C4a), 118.99 (C-4-purine), 118.26 (CH-aromatics), 122.10, 125.65, 127.29, 140.77 (CH-8-purine), 148.55 (C-4-purine), 151.07 (C-8a), 152.48 (CH-2-purine), 154.85 (C-6-purine). HR (LSIMS) m/z calcd for C15H16N5OS [M + H]+ 314.1076, found 314.1077.
(R)-N-Methyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(R)-24] (72 mg, 42%): ee = 99.0%, [α]25D −65.83 (c 1.0 in MeOH); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-24 = 26.679 min, tR (S)-24 = 29.495 min.
(S)-N-Methyl-9-(2,3-dihydro-1,4-benzoxathiin-3-ylmethyl)-9H-adenine [(S)-24] (72 mg, 42%): ee = 59.1%, [α]25D + 38.3 (c 1.0 in MeOH); HPLC analysis: hexane–EtOH = 80/20, flow rate 1.0 mL min−1, λ = 250 nm, tR (R)-24 = 26.576 min, tR (S)-24 = 28.695 min.
Conclusions
We have described herein a novel and efficient synthetic method between alkylated aminopurines (N,N-dimethyl-, 2-chloro-N-methyl-, and N-methyl-adenines) and the achiral and chiral secondary alcohol 3,4-dihydro-2H-1,5-benzoxathiepin-3-ol 12 by the Mitsunobu reaction under microwave-assisted conditions to obtain 17, 18, 22–24. We have confirmed that the sulfur atom of the seven-membered heterocycle exerts a remarkable neighboring group effect on its reaction with several amino-purines, producing a complete SN2 inversion at the stereogenic centre after the corresponding ring-contraction. Alkylation of purines usually results in the formation of N-9 and N-7 alkylpurines, the N-9 regioisomer normally being the major product.1–3 Nevertheless, N-3 regioisomers of purines are scarcely reported, especially when a chiral moiety is linked to the purine ring, and these substructures could lead to specific inhibitors of known biological targets and pave the way to new inhibitors or antagonists of targets that have yet to be described. Several biological studies on the chiral and achiral molecules described here are underway and the corresponding results will be communicated in due course.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by the Instituto de Salud Carlos III (Fondo de Investigación Sanitaria) through the project no. PI10/00592 and by the ERDF (European Regional Development Fund). The M. E. G.-R. FPU grant AP2007-02954 from the Ministerio de Ciencia e Innovación of Spain is greatly acknowledged. The project “Factoría de Cristalización” Consolider Ingenio-2010 (CSD2006-00015) provided X-ray structural facilities for this work.Notes and references
- M. C. Núñez, M. Díaz-Gavilán, A. Conejo-García, O. Cruz-López, M. A. Gallo, A. Espinosa and J. M. Campos, Curr. Med. Chem., 2008, 15, 2614 CrossRef.
- J. A. Marchal, M. C. Núñez, A. Aránega, M. A. Gallo, A. Espinosa and J. M. Campos, Curr. Med. Chem., 2009, 16, 1166 CrossRef CAS.
- C. López-Cara, A. Conejo-García, J. A. Marchal, G. Macchione, O. Cruz-López, H. Boulaiz, M. A. García, F. Rodríguez-Serrano, A. Ramírez, C. Cativiela, A. I. Jiménez, J. M. García-Ruiz, D. Choquesillo-Lazarte, A. Aránega and J. M. Campos, Eur. J. Med. Chem., 2011, 46, 249 CrossRef PubMed.
- O. Caba, M. Díaz-Gavilán, F. Rodríguez-Serrano, H. Boulaiz, A. Aránega, M. A. Gallo, J. A. Marchal and J. M. Campos, Eur. J. Med. Chem., 2011, 46, 3802 CrossRef CAS PubMed.
- A. Conejo-García, M. E. García-Rubiño, J. A. Marchal, M. C. Núñez, A. Ramírez, S. Cimino, M. A. García, A. Aránega, M. A. Gallo and J. M. Campos, Eur. J. Med. Chem., 2011, 46, 3795 CrossRef PubMed.
- O. Caba, F. Rodríguez-Serrano, M. Díaz-Gavilán, A. Conejo-García, P. Álvarez, R. Ortiz, A. Martínez-Amat, J. M. Campos, M. A. Gallo, J. A. Marchal and A. Aránega, Eur. J. Med. Chem., 2012, 50, 376 CrossRef CAS PubMed.
- A. M. Michelson, in The Chemistry of Nucleosides and Nucleotides, Academic Press, Inc., London, New York, 1963 Search PubMed.
- J. Reynolds and M. Kassiou, Curr. Org. Chem., 2009, 13, 1610 CrossRef.
- W. Li, X. Yin and S. W. Schneller, Bioorg. Med. Chem. Lett., 2008, 18, 220 CrossRef CAS PubMed.
- H. Kumamoto, N. Takahashi, T. Shimamura, H. Tanaka, K. T. Nakamura, T. Hamasaki, M. Baba, H. Abe, M. Yano and N. Kato, Tetrahedron, 2008, 64, 1494 CrossRef CAS PubMed.
- H. Li, J. C. Yoo and J. H. Hong, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 1238 CAS.
- J. A. Gómez, J. M. Campos, J. A. Marchal, M. A. Trujillo, C. Melguizo, J. Prados, M. A. Gallo, A. Aránega and A. Espinosa, Tetrahedron, 1997, 53, 7319 CrossRef.
- M. C. Núñez, M. G. Pavani, M. Díaz-Gavilán, F. Rodríguez-Serrano, J. A. Gómez-Vidal, J. A. Marchal, A. Aránega, M. A. Gallo, A. Espinosa and J. M. Campos, Tetrahedron, 2006, 62, 11724 CrossRef PubMed.
- M. Díaz-Gavilán, A. Conejo-García, O. Cruz-López, M. C. Núñez, D. Choquesillo-Lazarte, J. M. González-Pérez, F. Rodríguez-Serrano, J. A. Marchal, A. Aránega, M. A. Gallo, A. Espinosa and J. M. Campos, ChemMedChem, 2008, 3, 127 CrossRef PubMed.
- J. E. Huber, in Encyclopedia of Reagents for Organic Synthesis, ed. L. A. Paquette, Wiley, New York, 1995, vol. 4, p. 2326 Search PubMed.
- M. E. García-Rubiño, A. Conejo-García, M. C. Núñez, E. Carrasco, M. A. García, D. Choquesillo-Lazarte, J. M. García-Ruiz, M. A. Gallo, J. A. Marchal and J. M. Campos, Curr. Med. Chem., 2013, 20, 4924 CrossRef.
- M. B. Smith and J. March, in March's Advanced Organic Chemistry, John Wiley & Sons, Inc, New Jersey, 6th edn, 2007, p. 436 Search PubMed.
- D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420 CrossRef CAS.
- A. Zakrzewska, R. Gawinecki, E. Kolehmainen and B. Ośmialowski, Int. J. Mol. Sci., 2005, 6, 52 CrossRef CAS PubMed.
- M. Miyaki and B. Shimizu, Chem. Pharm. Bull., 1970, 18, 1446 CrossRef CAS.
- Y. Kitade, T. Ando, T. Yamaguchi, A. Hori, M. Nakanishi and Y. Ueno, Bioorg. Med. Chem., 2006, 14, 5578 CrossRef CAS PubMed.
- M. Yang, S. W. Schneller and B. B. Korba, J. Med. Chem., 2005, 48, 5043 CrossRef CAS PubMed.
- X. Yin, W. Li and S. W. Schneller, Tetrahedron Lett., 2006, 47, 9187 CrossRef CAS PubMed.
- M. Yang, W. Ye and S. W. Schneller, J. Org. Chem., 2004, 69, 3993 CrossRef CAS PubMed.
- N. B. Dyatkina, F. Theil and M. Von Janta-Lipinski, Tetrahedron, 1995, 51, 761 CrossRef CAS.
- M. S. Iyer, M. Palomo, K. M. Schilling, Y. Xie, L. Formanskii and D. E. Zembower, J. Chromatogr. A, 2002, 944, 263 CrossRef CAS.
- R. W. Taft Jr and I. C. Lewis, J. Am. Chem. Soc., 1959, 81, 5343 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Chromatograms and RMN studies of enantiomers of 12 and 13, and racemates and enantiomers of 17 and 22–24 are provided. CCDC 864711, 864709 and 864710. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra01968g |
| This journal is © The Royal Society of Chemistry 2014 |