Evaluating solvent extraction systems using metabolomics approaches†
Amanda C.
Martin
a,
Alison D.
Pawlus
a,
Erin M.
Jewett
a,
Donald L.
Wyse
b,
Cindy K.
Angerhofer
c and
Adrian D.
Hegeman
*a
aDepartment of Horticultural Science, Plant Biological Sciences Graduate Program, Department of Plant Biology, and Microbial and Plant Genomics Institute, University of Minnesota, 305 Alderman Hall, St Paul, MN 55108, USA. E-mail: hegem007@umn.edu
bDepartment of Agronomy and Plant Genetics, University of Minnesota, 411 Borlaug Hall, St. Paul, MN 55108, USA
cAveda Corporation, Blaine, MN 55449, USA
First published on 2nd June 2014
Abstract
Metabolic fingerprinting was performed on a set of botanical extracts to compare the extraction efficiency of different solvents to inform the construction of phytochemical libraries. We compared the extraction efficiency, examining both yield and chemical diversity, of eight single-solvent extractions prepared in parallel and using solvent–solvent partitioning. Three-dimensional data were reduced into features, which were used as unbiased metrics to identify solvents that would produce botanical extracts with the greatest chemical diversity. Chemical diversity and extract yield did not necessarily increase together. For each species and tissue, the total number of observable chemical features closely approached maximum values when three different single-solvent extractions were performed in parallel. The dynamic range of detectable compounds in plant extracts was increased significantly by performing solvent partitioning. Overall, maximum chemical diversity in a plant extract was most efficiently approached if solvent partitioning was performed on an extract made with 70% ethanol. We have shown that using metabolic fingerprinting is useful for assessing compound diversity in complex plant extracts.
1. Introduction
One might think, given the tremendous importance of plant natural products in medicine and commercial product formulation1,2 that broadly applicable procedures for natural product extraction would be well defined and firmly supported by methodological experimentation. It is surprising to see how little information is available concerning optimized extraction protocols that provide maximal chemical diversity in plant extracts given the theoretical importance of sampled chemical diversity for compound discovery through high-throughput screening approaches. Extracting natural products from plant material to funnel into high-throughput screens (HTS) is an effective strategy for testing a broad range of bioactivities nearly simultaneously. Through HTS valuable chemicals may be uncovered from libraries composed of extract fractions or pure compounds. Plant natural products have a wide variety of physicochemical properties and may be present across a huge range of concentrations. There are a number of different methods available for plant extractions, some requiring specialized equipment such as: supercritical fluid extraction, Soxhlet extraction, pressurized solvent extraction, microwave-assisted, steam/hydro-distillation, decoction, infusion, percolation, pressing, and boiling.3 Solvent impurities and their tendency to form artifacts, such as the condensation products formed with acetone, also must be considered during the selection of extraction solvents.4 Extraction requires efficient compound solubilization from a diverse set of plant tissue matrices making the optimization of generalized extraction protocols quite challenging. To date, this challenge has been the subject of many studies that attempt to determine ideal extraction conditions for detection of compounds by either monitoring a specific biological activity,5–10 a targeted compound class,10–16 or individual molecules.17 One consequence of these extraction optimization strategies is that they ultimately bias the chemical diversity of the resulting extracts toward whichever selection criteria were imposed. This is potentially damaging to the success of high-throughput chemical library screening approaches that depend on the availability of maximal compound diversity within screened populations. In this study, we have used an LC-MS metabolic fingerprinting approach in an attempt to minimize this bias in evaluating extract chemical diversity to enable a more inclusive assessment of chemical extraction efficiency. In so doing we acknowledge the usefulness of measurements of both known chemicals and the utility of observable yet unknown chemical entities for assessing total extract chemical diversity.Metabolomics, the comprehensive study of all small molecules within a biological system, includes the technique of metabolic fingerprinting, which when paired with multivariate statistical analysis (MVA), facilitates an examination of the global molecular diversity within a whole extract.18,19 Metabolic fingerprinting has been carried out using a variety of analytical platforms including liquid chromatography-mass spectrometry (LC-MS).20,21 LC-MS is particularly well suited for the analysis of botanical extracts because they are composed of diverse array of chemical species that range widely in concentration.17,21 In a metabolic fingerprinting experiment, individual chemical fingerprints are collected by LC-MS from replicate samples. Continuous LC-MS data are simplified into discrete sets of features for each sample through a process called data reduction. Each feature is made up of a unique retention time (tR), a monoisotopic mass, and a relative intensity value that varies from feature to feature from sample to sample and must be greater than zero counts.22 Thus, the total feature set provides a reasonable approximation of chemical composition of a sample without requiring the laborious process of chemical structural characterization for all of the sample's components. While some representational bias is present in LC-MS due to competition for ionization energy amongst coeluting chemical species, this bias will be significantly less than other common general analytical techniques such as NMR, which is much less sensitive, GC-MS or GC-FID, which require volatile analytes, or LC-UV/Vis, which requires the presence of a detectable chromophore in each analyte.23
Plant materials for this study were selected from a large number of local (Minnesota, USA) plant species shown to display antimicrobial and antioxidant activities in previous work.5,24,25 We used three of those plant species, Rhus typhina L. (staghorn sumac), Lythrum salicaria L. (purple loosestrife), and Monarda fistulosa L. (wild bergamot or bee-balm), to provide a diverse set of plant materials for this study that would, taken in toto, be fairly representative of plant materials in general. Multiple parallel single-solvent extractions and three-part extraction partitions using solvents of variable selectivity such as water, ethanol, and dichloromethane were compared.6,20 Extract concentrations were calculated and overall percent yields were determined from residual mass measurements following solvent evaporation. Chemical diversity was evaluated using metabolic fingerprinting by ultra-performance liquid chromatography-electrospray ionization-single quadrupole mass spectrometry (UPLC-ESI-SQ-MS) paired with MVA. Overall, we evaluated extract reproducibility, yield, and the number and uniqueness of detected metabolite features among the different extraction methods.
2. Experimental
2.1 Materials and reagents
2.2 Sample preparation
Two different sets of extraction experiments were performed on dry ground material (Fig. 1). Experiment one consisted of eight single solvent extractions performed in parallel and experiment two was a series of single solvent extractions followed by partitioning with hexanes and dichloromethane. Experiment one provided an initial assessment of eight solvents systems: hexanes, dichloromethane, ethyl acetate, methanol, isopropanol, water, aqueous ethanol (ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water, 70:30 v/v), and a dichloromethane–methanol mix (dichloromethane:methanol, 1:1 v/v). We used aerial tissue, consisting of stems, leaves, flowers, and buds, from L. salicaria and M. fistulosa; the set of extracts generated from a single species were compared with one another. Briefly, a recorded exact weight between 100–200 mg of dry ground plant material was placed into 2 mL polypropylene microcentrifuge tubes and 1.5 mL of solvent was added. The tubes were individually mixed using a Fisher Scientific fixed speed mini vortexer (Scientific Industries Inc. Bohemia, NY, USA) and then turbo-mixed using a Fisher Scientific vortex Genie 2™ (Scientific Industries Inc., Bohemia, NY, USA) for 15 min. This step was repeated and then the tubes were centrifuged using an Eppendorf 5415C centrifuge (Brinkman Instruments, Westbury, NY, USA) at 12000 rpm for 5 min. The extract supernatant was removed to a clean tube and placed at 4 °C in the dark. Each extraction was replicated 4 times.
:water, 70:30 v/v), and a dichloromethane–methanol mix (dichloromethane:methanol, 1:1 v/v). We used aerial tissue, consisting of stems, leaves, flowers, and buds, from L. salicaria and M. fistulosa; the set of extracts generated from a single species were compared with one another. Briefly, a recorded exact weight between 100–200 mg of dry ground plant material was placed into 2 mL polypropylene microcentrifuge tubes and 1.5 mL of solvent was added. The tubes were individually mixed using a Fisher Scientific fixed speed mini vortexer (Scientific Industries Inc. Bohemia, NY, USA) and then turbo-mixed using a Fisher Scientific vortex Genie 2™ (Scientific Industries Inc., Bohemia, NY, USA) for 15 min. This step was repeated and then the tubes were centrifuged using an Eppendorf 5415C centrifuge (Brinkman Instruments, Westbury, NY, USA) at 12000 rpm for 5 min. The extract supernatant was removed to a clean tube and placed at 4 °C in the dark. Each extraction was replicated 4 times.
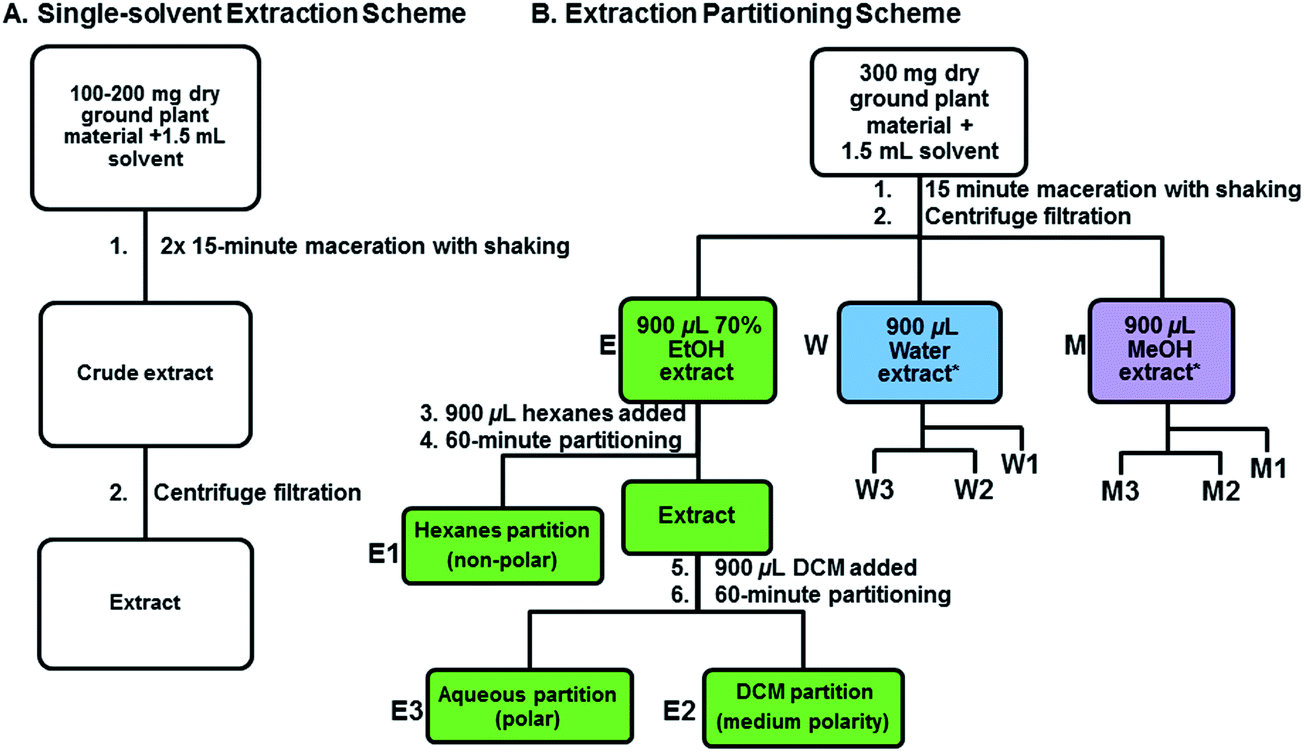 | ||
| Fig. 1 Schematic of workflows for (A) parallel single-solvent extractions and (B) extraction partitioning. Single solvent extractions with hexanes, dichloromethane, ethyl acetate, methanol, isopropanol, water, aqueous ethanol (ethanol:water, 70:30 v/v), or a dichloromethane–methanol mix (dichloromethane:methanol, 1:1 v/v) follow the simple linear workflow shown in (A). For the more complicated extract partitioning workflow shown in (B), 300 mg of dry plant material was extracted with 1.5 mL of either 70% ethanol in water (E, green), pure water (W, blue), or pure methanol (M, violet). Each of these extracts was then partitioned against an equal volume of first hexanes (E1, W1, M1), and then DCM (E2, W2, M2), which leaves a residual polar alcohol or aqueous phase (E3, W3, M3). *The complete schematic is illustrated in detail for the 70% ethanol in water extract partitioning (green boxes) and is abbreviated for the water (blue) and methanol (violet) extracts. | ||
Experiment two was a single-step extraction followed by two solvent partitioning steps using the most effective solvents from experiment one. We focused on three different tissue types from one species, namely, R. typhina leaves, berries, and stems. The single-step extraction was prepared using the same general method as in experiment one with methanol, 70% ethanol, dichloromethane, hexanes, water, and dichloromethane–methanol. The extract partitioning was performed on an initial extract prepared from 70% ethanol in water, 100% water, or 100% methanol, followed by a two-step partitioning with hexanes and then dichloromethane (Fig. 1B). The amount of starting material was increased to 300 mg, to ensure adequate quantities after partitioning, and the shaking was performed on a 2010 Geno/Grinder® (SPEX Sample Prep, Metuchen, NJ, USA) using a 15 min shaking program (5 min at 500 rpm followed by 10 min at 700 rpm). After centrifugation the prepared extract supernatant (about 900 μL) was removed to a clean tube and 900 μL of hexanes was added. After being mixed on the Geno/Grinder for 3 min at 700 rpm the two immiscible layers were allowed to separate for 60 min. After phase separation, the nonpolar hexanes layer (Fig. 1B: E1, W1, M1) was removed from the polar layer (Fig. 1B: E3, W3, M3) and 900 μL of dichloromethane was added to the polar layer for partitioning using the same procedure to generate a medium polarity partition (Fig. 1B: E2, W2, M2). All three partitions were separated into clean tubes, centrifuged at 12000 rpm for 5 min, and stored at 4 °C in the dark. All extract partitions were replicated 4 times.
2.3 Metabolic fingerprinting
3. Results and discussion
3.1 Extraction yield
The extraction efficiencies of the eight parallel single-solvent systems used to prepare extracts from L. salicaria and M. fistulosa aerial tissues were evaluated (Fig. 2). The six best solvents from experiment one were then used to extract R. typhina leaves, berries, and stems, and the extraction efficiencies were assessed (Fig. 3). All five sets of extracts show similar trends in the variation of overall percent yield, although absolute extract concentration differed greatly (Fig. 2 and 3). A visual inspection shows marked differences in the appearance of the extracts with 70% ethanol, methanol, and dichloromethane–methanol producing similar looking, dark extracts; water and dichloromethane producing slightly lighter and variably colored extracts, and isopropanol, ethyl acetate, and hexanes producing very light yellow extracts with hexanes being almost colorless (Fig. S1†). Extracts prepared with 70% ethanol and, despite its light appearance, water had the highest percent yield for all plant materials except for R. typhina berries and stems, where methanol produced higher yielding extracts than water. The extracting solvents, methanol, dichloromethane–methanol, dichloromethane, and hexanes produced moderately yielding extracts, although for R. typhina berries extracted by dichloromethane yields were high. Both isopropanol and ethyl acetate extracts had low yields of less than two percent for all plant materials. Although the trends in percent yield were similar among the solvent systems, the absolute concentration of the extracts varied greatly according to the type of botanical material used. Extract concentrations ranged from less than 2 mg mL−1 to greater than 40 mg mL−1, where extracts prepared from M. fistulosa aerial parts and R. typhina stem tissue had the lowest concentrations overall. This absolute difference in extract concentration is similar to results from a study by Johansen et al., where differences in overall yield were found when using an identical extraction method on field peas, toasted soybean meal, cotton seed meal, and a feed mixture.12 Therefore, it is important to test a range of raw materials when evaluating extraction efficiency. Due to the low yields of samples prepared with isopropanol and ethyl acetate, these two solvents were eliminated from the more in depth study with R. typhina leaves, berries, and stems where we performed single solvent extractions followed by partitioning to evaluate the advantages, if any, that partitioning would lend to chemical diversity or yield. | ||
| Fig. 2 Comparison of extraction efficiency for extracts prepared from 8 parallel single-solvent extractions using aerial tissue from (A) L. salicaria and (B) M. fistulosa. Bars show the average percent yield of 4 replicate extractions, as related to the initial dry weight of plant material used, the error bars represent standard error. Letter superscripts indicate statistically different groups at a p-value ≤0.01 according to Tukey's HSD test. The number of features detected from metabolic fingerprints of the different solvent extractions is displayed above the letter superscripts. | ||
 | ||
| Fig. 3 Comparison of extraction efficiency for extracts prepared from 6 parallel single-solvent extractions using Rhus typhina (A) leaf, (B) berry, and (C) stem tissue. Bars show the average percent yield of 4 replicate extractions, as related to the initial dry weight of plant material used, the error bars represent standard error. Letter superscripts indicate statistically different groups at a p-value ≤0.01 according to Tukey's HSD test. The number of features detected from metabolic fingerprints of the different solvent extractions is displayed above the letter superscripts. | ||
Conclusions drawn solely from yield measurements indicate that in decreasing order of efficiency, 70% ethanol, water, methanol, and dichloromethane–methanol are the most efficient extraction solvents. This result is similar to a study by Sultana et al., who found that aqueous organic extracts yielded higher than absolute organic extracts.10 While percent yield measures crude extraction efficiency in terms of quantity, it does not indicate extract chemical diversity. For instance, a solvent system may result in a high yielding extract of primarily tannins and sugars and, therefore, lack molecular diversity. Thus, a solvent that is high yielding may produce extracts of low overall quality due to limited chemical diversity.
3.2 Extract chemical diversity
The second aspect of extraction efficiency is chemical diversity. We have attempted to find a maximally inclusive way to assess this factor. Previous research has used biological activity,7,9 levels of specific classes of compounds (e.g. tannins, flavonoids, alkaloids, saponins, etc.), marker compounds (e.g. quercetin or emodin), or major constituents to evaluate extraction efficiency.10,14,15 These methods may bias results towards highly abundant common molecules rather than systematically evaluating the total extractable chemical diversity from each species and plant part.11,26 Metabolic fingerprinting favors the analysis of whole extract chemical diversity rather than single components or known compound classes, making it particularly suitable for the inclusion of unknowns when assessing extraction efficiency.3.3 Metabolic fingerprinting
The acquisition of LC-MS generated fingerprints provides a visual way to qualitatively evaluate chemical diversity.8 Chemical diversity can be quantified by applying MVA to LC-MS fingerprints to generate features that can be used as unbiased metrics for comparison. While collecting LC-MS fingerprints it is essential to maintain a high level of reproducibility among the factors describing a feature for any given sample, those factors being m/z, intensity, and tR. Reproducibility of the first two factors, m/z and intensity, are a function of the mass spectrometer. Using unidentified features as metrics made it possible to obtain nominal mass measurements with a single quadrupole instrument. Run-to-run tR reproducibility is also important. We designed an LC-MS gradient and wash cycle that would provide high peak capacity and tR reproducibility without being prohibitively long since metabolomics typically requires the acquisition of many repeated measurements.Although there has been a move towards using very short gradient times, other reports show that increasing the gradient time results in increased numbers of detected features and better separation, particularly later in the gradient; therefore we used a 20 min one-step gradient.27–29 Schellinger et al., showed that run-to-run retention time reproducibility can be achieved by using higher flow rates, higher initial solvent strength, and a 2 column volume (cv) wash.30 This very short wash provides an opportunity to reduce overall cycle time. We included a 2 min hold at initial conditions (2% B) to decrease the heterogeneity effect on early eluting compounds. We increased the number of re-equilibration cvs from the suggested 2 to 6 to compensate for our moderate flow rate and initial solvent composition, but remained well below the commonly used 10–15 cvs.30 This short 5 min re-equilibration is less than half our gradient time, facilitating the analysis of more samples per unit time. All solvent extractions were run using our optimized LC-MS parameters.
3.4 Principal components analysis
While LC-MS chromatograms provided a visually accessible overview of the chemical diversity, there is too much information to fully evaluate the chemical diversity of the extracts by manually examining chromatograms. By performing feature detection and MVA, it is possible to quantify extract chemical diversity, enabling an unbiased comparison among extracts. Each plant species has a unique set of chemicals, in terms of compound types and concentration, and this is reflected in the feature set for each set of extracts. Each feature set is made up of the total number of unique features reproducibly detected in a given set of extracts. Features had to be present in three out of four replicate samples to be included in the final feature set. Certain solvents generated extracts with fewer features than other solvents where the relative intensity of any given feature is zero counts. For example, hexanes extractions typically had more features with an intensity of zero than 70% ethanol extracts (Fig. 2 and 3). This reduction in detected features may be due to the narrower selectivity and poor cell penetration of hexanes resulting in extracts with fewer features. Alternatively, the low number of features observed for non-polar extracts prepared with hexanes or dichloromethane may also be partly attributable to the detection bias of RPLC-ESI-MS, which favors the detection of more polar metabolites.In general, we found 70% ethanol extracts to have the highest number of features for all plant material; this finding is in contrast to those in a study by Want et al., where methanol extracts of human serum had the highest feature numbers.29 Methanol has been shown to be very effective at precipitating proteins, a key factor for animal and human based metabolomics studies. Additionally, it has been shown that esterification reactions in the presence of methanol can degrade polyphenolics, saponins, and lipids in plant extracts.31,32 We acknowledge that isopropanol, a secondary alcohol, would result in fewer artifacts resulting from esterification reactions. However, the longer alkyl chain of ethanol provides a significant decrease in esterification rates over methanol. Using ethanol as an extracting solvent for plants has additional benefits including its low cost and usefulness in USDA certified “organic” food, medicinal, and cosmetic products.24 Even with the differences in sample types and optimal extraction solvent the size of the complete feature sets for the plant material evaluated, 3790 features for L. salicaria, 781 features for M. fistulosa; and 1645 for R. typhina leaves; 1378 for berries; and 1179 for stems were similar to the 2000 features detected in methanol–acetone extracts of human serum.29 The variation in the total number of features reflects the chemical variability of the different species and tissue types.
In addition to quantity, qualitative characteristics of features are also an important comparison metric. Visualizing how the features from a particular solvent extract are distributed across chromatographic and mass spectral space provides information about the elution time and mass of detected molecules. The feature set of L. salicaria, represented by black dots is well distributed across this space (Fig. 4). The distribution of features detected in each of the subsequent solvent extractions is also plotted. The extracts prepared from methanol, 1:1 dichloromethane:methanol, and 70% ethanol appear to contain features distributed most similarly to the entire feature set indicating that these solvents more completely extract the chemical diversity of the plant material. The feature distribution of extracts prepared with water is heavily weighted towards early retention times and small m/z, indicating that smaller, polar molecules were mostly present in these extracts. Conversely, hexanes and dichloromethane extracts have feature distributions that indicate the extraction of well-retained, non-polar molecules. Additionally, these two solvent extracts show a feature rich area centered around 20 minutes and m/z 700 that is largely absent from the other solvent extract feature distributions. Extracts prepared using isopropanol and ethyl acetate show a decreased number of features and an absence of any unique distribution coverage, further supporting the elimination of these two solvents from consideration for maximizing extraction efficiency.
 | ||
| Fig. 4 Scatterplots showing the distribution of features detected from metabolic fingerprints of extracts prepared from 8 parallel single-solvent extractions with L. salicaria aerial tissue. Individual features are represented by black dots and the total number of features detected from each solvent extract is listed parenthetically. This feature set consists of 3790 features in all. | ||
To aid in showing how solvent extracts are globally related to each other, a principal components analysis (PCA) was carried out. PCA plots display information about sample sets including: (1) LC/MS fingerprinting reproducibility through tight clustering of identical extractions, (2) similarity between different solvents via secondary groupings of clusters, and (3) where the secondary groupings form in relation to the principal components (PC) (Fig. 5). Extract clusters located away from the PCA plot origin are enriched in a particular set of metabolites. Predictions can be made about how secondary groupings might form based on prior knowledge of the chemical selectivity of the extraction solvents.
 | ||
| Fig. 5 Representative principal components analysis of L. salicaria parallel single-solvent extractions. Principal component (PC) 1 and 2 are shown and together account for 31.7% of the variation in this sample set. Additional PCs 3, 4, 5, and 6 explain 9.6%, 6.6%, 5.0% and 4.1% of the variation, respectively. No single principal component is able to capture a large amount of the variation of the whole set of extraction solvents because of the significant differences between each of solvents individually. Each solvent system has a large amount of highly reproducible unique parameters resulting in the tight cluster groups of replicate extracts produced with a single solvent; however the variation of the sample set as a whole cannot be easily mapped into a single PC. Over 50% of the variance is explained by the first 5 PCs. Secondary groupings I, II, and III are composed of replicate extract clusters that are similar to each other. | ||
The effect of the solvents on final extract chemical composition was significant, where PC 1 & 2 explained between 31 and 50% of the variation for all of the sample sets. This level of variation was driven solely by the extraction solvents. The highly reproducible and unique solvent parameters resulted in tight clusters of replicate extracts prepared with a single solvent. A pairwise plot of PC 1 & 2 for the L. salicaria sample set shows the distribution of replicate extract clusters forming secondary groups that separate well across both PCs to form three distinct groups (Fig. 5). Group I contains extracts prepared with methanol, 70% ethanol, and water; Group II contains extracts prepared with hexanes, isopropanol, ethyl acetate, and dichloromethane; and extracts prepared with the dichloromethane–methanol mix form an isolated Group III. The isolated location of the dichloromethane–methanol extract indicates that it contains unique features that are either not present or at undetectable levels in the other extracts. These secondary grouping trends are common among all sets of extracts from all plants and plant parts. The formation of the secondary groupings is reasonable based on the similarities and differences among the solvents used. The plot provided a quick method to visualize and compare the chemical diversity of the different solvent extractions. Typically, the next step would be to use the PC loadings plot to identify the main features responsible for the variation in the different extraction conditions.33 Here, however, we continued to use the entire feature-set to assess whole extract chemical diversity.
3.5 Feature comparison
Our aim was to find a set of solvents used to prepare separate single-solvent extracts in parallel that would approach maximum chemical diversity for any plant material. Metabolomics fingerprinting experiments generate large datasets that approach a thorough characterization of whole extract chemical diversity. An examination of the features from any two separately prepared parallel single-solvent extractions enables a comparison of both unique and shared features between the two (Fig. 6a). Increases in chemical diversity are seen when examining combinations of two parallel extractions that have the tallest overall bar, which corresponds to the greatest total number of features. This bar will also have the smallest gray shaded area, which corresponds to a lower number of shared features. The total number of features and number of shared features differ greatly, depending on which two solvent extractions are compared with one another (Fig. 6a). Two-solvent combinations that include hexanes, ethyl acetate, or isopropanol have fewer total features. Increasing the number of solvent extractions results in a greater total number of features; however, this increase begins to level off when feature sets from three parallel single solvents extracts are compared to each other (Fig. 6b). Although only the analysis of L. salicaria extracts is shown, the other extract sets behaved remarkably similarly in that the maximum number of features is approached when three different solvent systems are used to extract one type of plant material. | ||
| Fig. 6 Comparison of extraction efficiency via chemical complexity for combinations of two and three parallel single-solvent extractions. The total numbers of unique observed features from L. salicaria aerial tissue single solvent extractions are shown for combinations of two different solvents (in A) and three different solvents (in B). The total numbers of features unique to a particular combination of solvent extractions are shown by the heights of each bar. The fraction of features uniquely found in a single solvent from each combination is shown by each colored bar segment (shaded according to the key). The fraction found in two (or more) solvents in each combination is indicated by the gray segment of each bar. All possible pairwise comparisons of the eight solvents are shown in (A). | ||
With the combinations of three parallel solvent extractions it is important to note which ones have the smallest area of shared features. It is these combinations that will maximize chemical diversity while having to perform the least number of parallel extractions. Our results suggest that the most promising three solvent combinations include: 70% ethanol, dichloromethane–methanol, and dichloromethane; 70% ethanol, hexanes, and dichloromethane–methanol; methanol, hexanes, and water; or 70% ethanol, water, and dichloromethane (Fig. 6b). Each of the resulting extracts fell into separate PCA secondary groupings, indicating that they contain different features (Fig. 5). The inclusion of 70% ethanol in the most efficient combinations is convenient because although flammable, ethanol is relatively safe, readily available, typically of high purity, completely biodegradable, and suitable for use in U.S.D.A. certified organic products.3
3.6 Extract partitioning
Solvent partitioning has been previously employed for plant extract screening programs.34 Partitioning separates polar and non-polar compounds to: reduce bioassay interferences, increase relative concentrations of minor compounds to detectable and/or active levels, decrease the prevalence of hydrophobic compounds contaminating chromatography columns, and simplify later compound isolation efforts.34 Using the same metric to evaluate extract efficiency, initial extractions with water, 70% ethanol, or methanol of the leaf, berry, and stem tissue from R. typhina were subjected to solvent partitioning with hexanes followed by dichloromethane. A visual inspection of the extract partitions shows that the aqueous partition from the methanol or 70% ethanol extracts have a similar green color, whereas the water generated extracts appear brown; also notable is the colorless appearance of the hexanes and dichloromethane partitions from an initial water extraction (Fig. S1b†).Feature detection on the extract partitions revealed an interesting trend. When either 70% ethanol or methanol was the initial extracting solvent, hexanes and dichloromethane partition feature numbers surpassed those of the single-solvent extractions with hexanes or dichloromethane (Fig. 7). Moreover, total number of unique features was higher for extract partitions than for single-solvent extraction combinations including hexanes, dichloromethane, and 70% ethanol or methanol. In R. typhina leaves, for instance, the number of unique features present in dichloromethane single solvent extraction was 40; the number of unique features in a dichloromethane partition of an initial 70% ethanol extract was 201. Similarly, the number of unique features in a hexanes extraction of R. typhina leaves was 25 compared to 70 unique features in the hexanes partition of an initial 70% ethanol extraction. This increase in feature number may be due to enrichment of low abundance metabolites in partitions where they are most soluble, raising their relative concentration to detectable levels.6,34 Additionally, compounds subject to ion suppression in the 70% ethanol extract may have ionized better when concentrated in the dichloromethane or hexanes partition resulting in their detection and inclusion as features. Overall, this increased access to low abundance compounds extends the dynamic range of detection methods and biological assays. Generally, this partition advantage was most strongly observed when 70% ethanol was the initial extracting solvent. Although, the total number of features resulting from hexanes and dichloromethane partitioning of an initial extract made with methanol was greater than the total number of features resulting from parallel single-solvent extractions with hexanes, dichloromethane, and methanol, both of these scenarios showed decreased overall feature numbers than when 70% ethanol was replaced by methanol as an extracting solvent. This increase in feature number was the case when 70% ethanol was the initial extraction solvent that was partitioned with hexanes and dichloromethane or if the three solvents were used in parallel single-solvent extractions (Fig. 7). Furthermore, when water was used as the initial extracting solvent, the number of features in both the hexanes and dichloromethane partitions were greatly reduced when compared to the number of features detected from a single-solvent extraction with either organic solvent. These results are similar to previous studies evaluating extracting solvents, which found 100% water to be inferior to methanol or ethanol in total quantity and diversity of compounds in an extract.12,14,29 One additional factor to consider is the use of polypropylene tubes for all extractions, if possible glass vials would be preferred as they would reduce potential for artifact formation, particularly with solvent mixtures containing dichloromethane. For consistency in this study polypropylene vials were used for all extractions and we recognize this as a study limitation. However, extracts prepared with dichloromethane did not have significantly increased feature numbers, plasticizers, or obvious signs of polymeric materials so we are confident that the potential formation of artifacts did not significantly bias the study results.
 | ||
| Fig. 7 Comparison of extraction efficiency via chemical complexity of combinations of three parallel single-solvent extractions and extraction partitions of R. typhina leaf, berry, and stem tissue. The total numbers of features unique to a particular combination of solvent extractions are shown by the heights of each bar. The fraction of features uniquely found in a single solvent or partition from each combination is shown by each colored bar segment (shaded according to the key). The fraction found in two (or more) solvents or partitions in each combination is indicated by the dark gray segment of each bar. The chemical complexity of three independent single-solvent extractions using 70% ethanol, hexanes, and dichloromethane is directly compared against solvent partitions generated from an initial extracting solvent of 70% ethanol followed by partitioning with hexanes and dichloromethane to generate partitions E1 (hexanes), E2 (dichloromethane), and E3 (polar alcohol) partitions (in A). The same comparison is shown in (B), where methanol replaces 70% ethanol. | ||
Although it seems possible to approach maximum chemical diversity by performing three parallel single-solvent extractions, there is an advantage to performing extract partitioning in certain situations. There was a strong increase in chemical diversity (number of features), but not yield, for non-polar solvents (e.g. hexanes, dichloromethane) that were part of a liquid–liquid, partitioning step when an alcohol, but not water, was used to perform the initial extraction (Fig. 7). The partition step, performed on the 70% ethanol extract, sufficiently enriched certain metabolites soluble in particular phases. This concentration effect may translate to detectable biological activity where there may not have been any previously. We generated pairwise plots of PC 1 & 2 for R. typhina extraction partitions and single-solvent extracts prepared from hexanes (Fig. S2†), dichloromethane (Fig. S3†), and water, 70% ethanol, and methanol (Fig. S4†). In all cases, the single-solvent extract clusters formed different secondary groupings from the extraction partitions indicating that partitioning of an initial extract effectively changes it. For example, hexanes and dichloromethane partitions of 70% ethanol or methanol extracts formed nearly overlapping secondary groupings that were more similar to their respective single-solvent extracts along PC 1, but differed along PC 2 indicating a difference in chemical composition between the single-solvent extract and partitions (Fig. S2 and S3†). When plotted together with the single-solvent extract prepared with water, the 70% ethanol or methanol single-solvent extracts formed an overlapping secondary grouping that was separated from the polar extract cluster, polar partition of a water extract, and polar partitions of 70% ethanol and methanol extracts, which also formed a secondary grouping (Fig. S4†).
4. Conclusions
A complete assessment of extraction efficiency comprises measurements of both extract yield and chemical diversity (Fig. 2 and 3). This study shows that the dynamic range of detectable compounds in a plant extract can be increased significantly by performing solvent partitioning. This increase in detectable compounds equates to an observed increase in chemical diversity. No single solvent extract can provide a complete feature set (Fig. 4), so maximum chemical diversity requires parallel single-solvent extractions with multiple solvent systems. In general, a good starting point for selecting a solvent is to choose one that contains greater than 50% of the total number of features distributed evenly over the chromatographic and mass spectral space and has greater than 5% yield.It is important to consider that low chemical diversity solvent extracts (those having smaller feature numbers) may still provide novel detectable bioactivities in some high throughput screens. For example, extracts generated from hexanes showed unique distributions patterns of features indicating the presence of different subsets of chemical entities (Fig. 4). If multiple parallel single-solvent extractions are to be performed, solvents from separate secondary groupings on the PCA plot, or those with unique distributions of features should be used in combination. Performing extract partitioning enhances the relative concentrations of low abundance compounds to detectable and potentially active levels, greatly extending the dynamic range of detection methods and biological assays. Obtaining maximum chemical diversity in a plant extracts is most efficiently approached if solvent partitioning is performed using an extract made with 70% ethanol or a comparable high efficiency solvent system (Fig. 7).
Using metabolomics-generated features provided a way to more globally assess the chemical diversity of plant extracts. A set of extraction parameters has been defined that can be used to build a phytochemical library that will approach a complete sampling of the chemical diversity contained in raw plant material. Metabolomics generated datasets are very large; we were able to make use of this large amount of information to thoroughly characterize the samples, without the need for rigorous metabolite identification. Metabolic fingerprinting combined with feature detection and MVA is a tool for global analysis that has the potential to be applied for quick evaluation of whole botanical extracts in initial chemical screens, aiding in natural product dereplication efforts and/or for quality control.
Acknowledgements
The authors would like to thank the botanicals research team in Estée Lauder R&D for helpful discussions, Stephen Brockman and Mary Musielewicz for technical assistance, and the Minnesota Supercomputing Institute (UMN MSI) at the University of Minnesota for software and computational support. This work was funded by the NSF Plant Genome Research Program grants IOS-0923960 and IOS-1238812, the NSF Graduate Research Fellowship Program (00006595), and the UNCF/Merck Science Initiative (ACM).Notes and references
- J. W. H. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed.
- N. Balasundram, K. Sundram and S. Samman, Food Chem., 2006, 99, 191–203 CrossRef CAS PubMed.
- F. Chemat, M. A. Vian and G. Cravotto, Int. J. Mol. Sci., 2012, 13, 8615–8627 CrossRef CAS PubMed.
- K. Hunchak and I. H. Suffet, J. Chromatogr., 1987, 392, 185–198 CrossRef CAS.
- P. Gillitzer, A. C. Martin, M. Kantar, K. Kauppi, S. Dahlberg, D. Lis, J. Kurle, C. Sheaffer and D. L. Wyse, J. Med. Plants Res., 2012, 6, 938–949 Search PubMed.
- W. P. Jones and A. D. Kinghorn, in Methods in Molecular Biotechnology™ Natural Products Isolation, ed. D. Sarker, Z. Latif and A. I. Gray, Humana Press, New Jersey, 2nd edn, 2006, ch. 13, pp. 323–352 Search PubMed.
- H. N. B. Marzoug, M. Romdhane, A. Lebrihi, F. Mathieu, F. Couderc, M. Abderraba, M. L. Khouja and J. Bouajila, Molecules, 2011, 16, 1695–1709 CrossRef PubMed.
- A. Nostro, M. P. Germanò, V. D'angelo, A. Marino and M. A. Cannatelli, Lett. Appl. Microbiol., 2000, 30, 379–384 CrossRef CAS.
- P. Singariya, K. K. Mourya and P. Kumar, J. Pharm. Sci. Res., 2011, 3, 1387–1393 CAS.
- B. Sultana, F. Anwar and M. Ashraf, Molecules, 2009, 14, 2167–2180 CrossRef CAS PubMed.
- C. W. Huie, Anal. Bioanal. Chem., 2002, 373, 23–30 CrossRef CAS PubMed.
- H. N. Johansen, V. Glitsø and K. E. Bach Knudsen, J. Agric. Food Chem., 1996, 44, 1470–1474 CrossRef CAS.
- R. Julkunen-Tiitto and S. Sorsa, J. Chem. Ecol., 2001, 27, 779–789 CrossRef CAS.
- B. Lapornik, M. Prošek and A. G. Wondra, J. Food Eng., 2005, 71, 214–222 CrossRef PubMed.
- F. Muanda, D. Koné, A. Dicko, R. Soulimani and C. Younos, Evid. Based Complement. Alternat. Med., 2011, 674320, 1–8 Search PubMed.
- J. Martinis, F. Kessler and G. Glauser, Plant Methods, 2011, 7, 23 CrossRef CAS PubMed.
- J. Saric, E. J. Want, U. Duthaler, M. Lewis, J. Keiser, J. P. Shockcor, G. A. Ross, J. K. Nicholson, E. Holmes and M. F. M. Tavares, Anal. Chem., 2012, 84, 6963–6972 CrossRef CAS PubMed.
- O. Fiehn, Plant Mol. Biol., 2002, 48, 155–171 CrossRef CAS.
- A. D. Hegeman, Briefings Funct. Genomics, 2010, 9, 139–148 CrossRef CAS PubMed.
- J. W. Allwood and R. Goodacre, Phytochem. Anal., 2010, 21, 33–47 CrossRef CAS PubMed.
- H. K. Kim and R. Verpoorte, Phytochem. Anal., 2010, 21, 4–13 CrossRef CAS PubMed.
- J. Trygg, E. Holmes and T. Lundstedt, J. Proteome Res., 2007, 6, 469–479 CrossRef CAS PubMed.
- K. Dettmer, P. A. Aronov and B. D. Hammock, Mass Spectrom. Rev., 2007, 26, 51–78 CrossRef CAS PubMed.
- J. R. Borchardt, D. L. Wyse, C. C. Sheaffer, K. L. Kauppi, R. G. Fulcher, N. J. Ehlke, D. D. Biesboer and R. F. Bey, J. Med. Plants Res., 2008a, 2, 81–93 Search PubMed.
- J. R. Borchardt, D. L. Wyse, C. C. Sheaffer, K. L. Kauppi, R. G. Fulcher, N. J. Ehlke, D. D. Biesboer and R. F. Bey, J. Med. Plants Res., 2008, 2, 98–110 Search PubMed.
- I. Raskin, D. M. Ribnicky, S. Komarnytsky, N. Ilic, A. Poulev, N. Borisjuk, A. Brinker, D. A. Moreno, C. Ripoll, N. Yakoby, J. M. O'Neal, T. Cornwell, I. Pastor and B. Fridlender, Trends Biotechnol., 2002, 20, 522–531 CrossRef CAS.
- P. A. Guy, I. Tavazzi, S. J. Bruce, Z. Ramadan and S. Kochhar, J. Chromatogr. B: Biomed. Sci. Appl., 2008, 871, 253–260 CrossRef CAS PubMed.
- A. Nordström, G. O'Maille, C. Qin and G. Siuzdak, Anal. Chem., 2006, 78, 3289–3295 CrossRef PubMed.
- E. J. Want, G. O'Maille, C. A. Smith, T. R. Brandon, W. Uritboonthai, C. Qin, S. A. Trauger and G. Siuzdak, Anal. Chem., 2006, 78, 743–752 CrossRef CAS PubMed.
- A. P. Schellinger, D. R. Stoll and P. W. Carr, J. Chromatogr., 2005, 1064, 143–156 CrossRef CAS PubMed.
- A. Tava, M. Mella, Z. Bialy and M. Jurzysta, J. Agric. Food Chem., 2003, 51, 1797–1800 CrossRef CAS PubMed.
- R. L. Lindroth and M. S. Pajutee, Oecologia, 1987, 74, 144–148 CrossRef.
- B. P. Bowen and T. R. Northen, J. Am. Soc. Mass Spectrom., 2010, 21, 1471–1476 CrossRef CAS PubMed.
- F. E. Koehn, in Progress in Drug Research, ed. F. Petersen and R. Amstutz, Birkhäuser Verlag, Basel, 2008, vol. 65, ch. 5, pp. 176–210 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02731k |
| This journal is © The Royal Society of Chemistry 2014 |