 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular characterization of an end-residue of humeomics applied to a soil humic acid†
A.
Nebbioso
a,
A.
Piccolo
*a,
M.
Lamshöft
b and
M.
Spiteller
b
aCentro Interdipartimentale di Ricerca per la Spettroscopia di Risonanza Magnetica Nucleare per l'Ambiente, l'Agro-alimentare e i nuovi Materiali (CERMANU), Università di Napoli Federico II, Portici, Italy. E-mail: alessandro.piccolo@unina.it
bInstitut für Umweltforschung, Dortmund Technische Universität, Otto-Hahn Str. 6, Campus North, D-44227 Dortmund, Germany
First published on 2nd May 2014
Abstract
Humeomics encompasses step-wise chemical fractionation and instrumental determination to fully characterize the heterogeneous molecular composition of natural organic matter. Humeomics applied to a terrestrial humic acid produced a recalcitrant end-residue, that was solubilized in alkali and size-fractionated by preparative high performance size exclusion chromatography (HPSEC). The ten separated size-fractions were then subjected to analytical HPSEC hyphenated with a high-resolution electrospray mass spectrometer (ESI-MS). Total ion chromatograms of size-fractions showed two eluting peaks, the molecular masses of which were identified with empirical formulae by ESI-MS. Most empirical formulae were easily associated with linear alkanoic, unsaturated, hydroxylated and hydroxy-unsaturated acids, as well as cyclic acids, but some compound structures were unclear. Tandem MS fragmentation was applied to one linear and three cyclic compounds, to elucidate their structures as hydroxyunsaturated hexanoic acid, two furane rings and a norbornane-like ring, respectively. The latter three compounds were never reported for terrestrial humic acid, but they resembled the carboxyl-rich alicyclic molecules, which had been proposed previously for dissolved organic matter. Quantitative measurement of components indicated that long-chain saturated acids were present in large-sized fractions more than in short-chain homologues, whereas unsaturated, hydroxylated and most cyclic acids were more abundant in small-sized fractions. This suggests that long, saturated and unsubstituted linear acids allow formation of large suprastructures, probably as a result of favorable intermolecular packing, compared with the irregularly shaped cyclic, unsaturated or hydroxylated compounds. We showed that humeomics clarifies the molecular composition and conformational arrangement of natural organic matter, and may help to elucidate the relationship between humic structure and environmental activity.
1. Introduction
Natural organic matter or humic substances (HS) refer to a class of naturally occurring organic compounds commonly found in soils, sediments and natural bodies.1 These substances play a pivotal role in environmental physical-chemistry and biology, and have great influence on soil conservation, quality and fertility.2,3 The heterogeneous and complex nature of HS makes it difficult to obtain definite and rigorous structural characterization of their molecular components. A recent view is that HS is composed of relatively low molecular-weight compounds organized in a supra-molecular structure held together by weak intermolecular bonds.4,5 According to this view, a thorough characterization of single components can be achieved only if they are adequately separated before analysis. A strategy based on step-wise chemical fractionation has proved highly efficient in reducing humic chemical complexity.6 Just as advances in biochemistry have resulted in modern genomics, advances in the understanding of humic chemical nature have resulted in novel separation-characterization methods of HS, which inspired the definition “humeomics”, a term introduced in analogy with other “omic” sciences.6 Moreover, application of size-exclusion chromatography to HS preliminarily separated into size-fractions has been shown to enhance analytical identification of humic molecules.7,8 Similarly, application of humeomics to size-fractions obtained by fractionating a bulk humic acid resulted in much better molecular identification for the size-separates than for the bulk material.9Key points of humeomics are (i) extensive fractionation of starting materials and (ii) minimal rearrangement of carbon backbone of native HS. The former ensures an increase in analytical yield,6,9 and the latter prevents formation of artifacts. This is an important step forward with respect to older destructive methods such as KMnO4 oxidation,10 which have been criticized because of resultant modification of the HS carbon backbone.11 Humeomics minimizes such a drawback by preferring reactions that do not affect C–C bonds, such as hydrolysis of ester and ethers groups.6
Although previous works succeeded in describing the type and amount of molecules separated in both organic solvents and aqueous media during humeomics, a humic fraction remained non-extractable at the end of the stepwise procedure, thereby preventing the complete detailed molecular characterization of humic matter constituents.6,9 The aim of this work was to solubilize any recalcitrant end-product of humeomics, decrease complexity of the method further by separating different size-fractions, and characterize the resulting molecular components by high-resolution electrospray mass spectrometry (ESI-MS).
2. Results and discussion
2.1 Preparative HPSEC separation
As the final end-product of humeomics, the humic residue RES4 is expected to be significantly less heterogeneous than the starting humic acid (HA). In fact, RES4 was depleted of humic components solubilized as unbound molecules in solvent extraction or after hydrolysis of ester or ether bonds. NMR and MS characterization showed such separated molecules to be prevalently composed of aliphatic and hydroxyl-alkyl compounds.6 Thus, RES4 residue became enriched in aromatic compounds with consequent enhancement of its chromophoric character. The increased aromaticity in RES4 was confirmed by CPMAS spectra, which showed a greater content of sp2 carbon (110–160 ppm) than the original HA and previous RES products of humeomics.6The enhanced light-absorbing capacity of RES4 caused by enrichment in sp2 carbons was revealed by the UV-detected preparative HPSEC chromatogram, with an elution profile showing three diffuse absorptions at around 28, 35 and 47 min of elution time. Such distribution of humic matter over the preparative HPSEC column allowed separation and collection of ten different size-fractions (Fig. S1 of ESI†). The greatest intensity in the elution profile resulted in the largest amount of humic matter in the corresponding size-fractions. In fact, the maximum intensity at 35 min (Fig. S1 of ESI†) yielded most of the eluting humic mass, as shown by the quantity obtained for the combined 2–4 size-fractions (Table S1 of ESI†).
2.2 Analytical HPSEC-ESI-Orbitrap-MS
The intensity of the two major signals varied greatly in the different TIC of RES4 and its ten size-fractions (Fig. S2 of ESI†). The first signal (20–24 min) progressively decreased when going from the largest-size to the smallest-size fraction, whereas the second signal (26–30 min) showed an opposite trend by steadily increasing its intensity with decreasing fractions size. This behavior indicates that the preparative size-fractionation of RES4 correctly separated humic fractions according to their hydrodynamic volume. Nevertheless, the change in signals intensity observed over the size-fractionation, suggests that the two TIC signals could have different distributions of large- and small-sized humic associations.
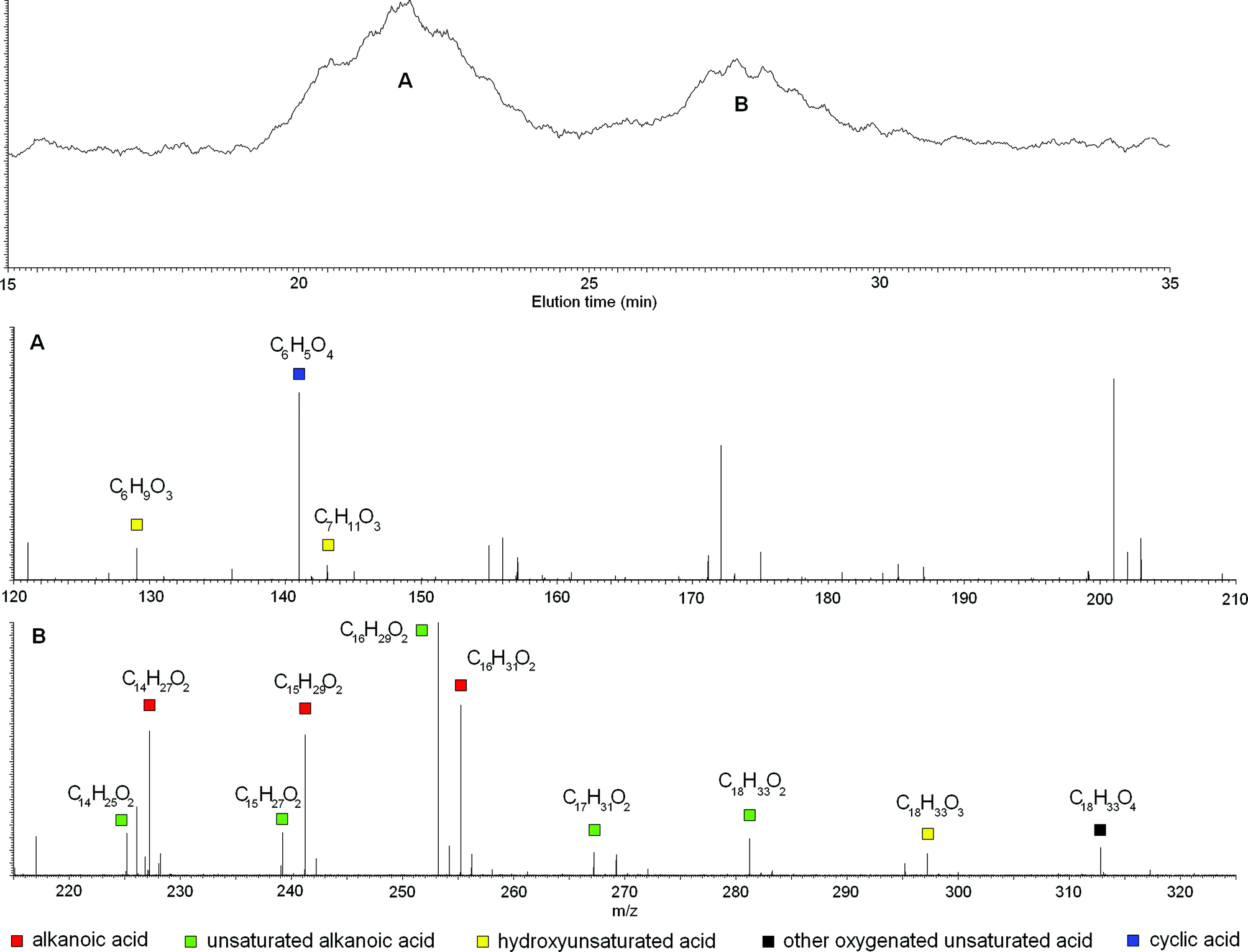 | ||
| Fig. 1 Mass peaks and related empirical formulae under two main peaks in HPSEC-ESI-MS elutions for bulk RES4. | ||
Other empirical formulae identified under TIC signals still suggested the presence of linear molecules but with less unsaturation and greater oxygenation than alkanoic acids. Double bonds together with oxygen-rich functions, such as carboxyl or hydroxyl groups, appeared concomitantly present in these molecules, although it was impossible to define their mutual substitution pattern from empirical formulae only. For example, the C18H33O4 formula that describes a mass eluted under the second TIC signal (Fig. 1B), may be attributed to either a saturated dicarboxylic acid or an unsaturated dihydroxy acid, and was therefore non-specifically defined as unsaturated oxygenated acid.
It is noteworthy that the molecular masses of identified empirical formulae did not follow the expected order of SEC elution time, by which analytes with large hydrodynamic radii elute before those with small radii (Table S2 of ESI†). In fact, molecules with formulae such as C7H6O8 (most likely a cyclic compound), C6H10O3 (a hydroxy-hexenoic acid), and C7H12O3 (a hydroxy-heptenoic acid), were eluted before most other molecules with empirical formulae of larger masses. Nevertheless, probable cyclic structures having small masses were eluted much faster (Table S2 of ESI†). An explanation of this contradictory behavior resides in the association of humic molecules in large heterogeneous conformations, the size of which and, thus, consequent HPSEC elution time, depends on the reciprocal arrangement of specific molecular structures.
A quantitative evaluation of empirical formulae related to other unsaturated and oxygenated acids, suggested a general progressive increase of unsaturation and oxygenation with decreasing size of fractions (Table 1). In fact, most of these molecules were less abundant in size-fractions separated at short rather than long elution times. For example, a compound with a C17H26O4 empirical formula was found to be only 13.3, 60.2, and 20.9 μg g−1 of total HA in the first three size-fractions, respectively, whereas it was found to be as much as 162.5, 153.8 and 110.8 μg g−1 of total HA in the last three size fractions (8–10, respectively) (Table 1). Such significant, although non-linear, increase of unsaturated oxygenated acids in small-size fractions suggests that these compounds were hardly stabilized in the large-size supramolecular associations eluting at earlier chromatographic time. This is expected to be a result of the poor associative capacity of such irregularly shaped carbon chains, as unsaturation and oxygen substitution rendered intermolecular aggregation more difficult than for the more hydrophobic unsubstituted alkanoic acids. In fact, the regular shape of the latter compounds favorably enabled their accommodation into larger size-fractions because of easier intermolecular packing in a heterogeneous suprastructure.
| Empirical formula | Bulk RES4 | Size-fractions | Sum of fractions | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |||
| Alkanoic acids | ||||||||||||
| C10H20O2 | 77 | 18 | 23 | 27 | 35 | 28 | 34 | 31 | 37 | 45 | 201 | 479 |
| C12H24O2 | 48 | 27 | 29 | 35 | 47 | 33 | 83 | 54 | 40 | 61 | 103 | 512 |
| C14H28O2 | 179 | 256 | 298 | 289 | 146 | 187 | 667 | 484 | 266 | 343 | 607 | 3544 |
| C15H30O2 | 191 | 389 | 411 | 317 | 431 | 230 | 791 | 545 | 229 | 372 | 400 | 4113 |
| C16H32O2 | 324 | 1000 | 1190 | 655 | 908 | 341 | 784 | 896 | 450 | 468 | 1090 | 7782 |
| C17H34O2 | 25 | 144 | 242 | 98 | 132 | 57 | 179 | 143 | 53 | 96 | 134 | 1278 |
| C18H36O2 | 8 | 122 | 145 | 104 | 422 | 25 | 67 | 160 | 50 | 59 | 99 | 1254 |
| Unsaturated alkanoic acids | ||||||||||||
| C14H26O2 | 46 | 37 | 52 | 64 | 80 | 36 | 168 | 106 | 53 | 98 | 117 | 812 |
| C16H30O2 | 326 | 537 | 708 | 828 | 1032 | 518 | 2054 | 1290 | 496 | 1035 | 899 | 9397 |
| C18H34O2 | 59 | 341 | 713 | 423 | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 108 108 |
168 | 507 | 415 | 150 | 678 | 625 | 17127 |
| Unsaturated hydroxyacids | ||||||||||||
| C6H10O3 | 33 | 15 | 24 | 24 | 38 | 58 | 34 | 65 | 32 | 46 | 176 | 511 |
| C7H12O3 | 23 | 17 | 28 | 13 | 33 | 35 | 38 | 42 | 27 | 21 | 93 | 346 |
| C18H34O3 | 22 | 46 | 21 | 50 | 72 | 29 | 49 | 41 | 18 | 106 | 243 | 675 |
| Other unsaturated oxygenated acids | ||||||||||||
| C15H22O4 | 9 | 22 | 24 | 12 | 73 | 67 | 186 | 368 | 71 | 217 | 426 | 1467 |
| C17H26O4 | 13 | 60 | 21 | 44 | 37 | 41 | 59 | 450 | 163 | 154 | 111 | 1138 |
| C17H26O5 | 19 | 12 | 9 | 6 | 40 | 36 | 65 | 233 | 59 | 103 | 80 | 642 |
| C18H34O4 | 22 | 9 | 5 | 9 | 13 | 3 | 14 | 12 | 6 | 23 | 16 | 110 |
| C18H36O4 | 4 | 33 | 8 | 8 | 43 | 2 | 7 | 5 | 3 | 22 | 27 | 157 |
| C18H34O5 | 4 | 31 | 4 | 3 | 125 | 2 | 1 | 3 | 2 | 51 | 34 | 257 |
| C19H30O5 | 2 | 28 | 10 | 8 | 10 | 19 | 23 | 391 | 9 | 103 | 12 | 611 |
| C19H28O4N2 | 0 | 7 | 0 | 0 | 5 | 0 | 0 | 0 | 2 | 7 | 4 | 26 |
| C19H30O6 | 8 | 7 | 7 | 2 | 23 | 25 | 54 | 256 | 26 | 125 | 52 | 576 |
| C20H24O3 | 9 | 25 | 21 | 8 | 6 | 4 | 8 | 24 | 6 | 29 | 30 | 161 |
| C21H26O3 | 8 | 19 | 16 | 3 | 4 | 2 | 5 | 12 | 3 | 15 | 16 | 94 |
| C21H34O6 | 0 | 15 | 3 | 3 | 3 | 12 | 8 | 265 | 4 | 67 | 7 | 385 |
| Cyclic acids | ||||||||||||
| C7H6O2 | 4336 | 16336 |
17068 |
19570 |
10345 |
9587 | 12093 |
14422 |
47210 |
12619 |
34382 |
193632 |
| C6H4O5 | 7888 | 27484 |
9568 | 54330 |
28015 |
19636 |
23654 |
24775 |
55316 |
18970 |
53960 |
315708 |
| C7H6O8 | 9158 | 11842 |
9711 | 65224 |
16093 |
9249 | 9092 | 7834 | 8826 | 5427 | 3518 | 146816 |
Similarly, the content of cyclic acids progressively increased with decreasing fraction size (Table 1). The corresponding C7H6O2 and C6H4O5 empirical formulae accounted, respectively, for 4336 and 7888 μg g−1 of total HA in fraction 1, whereas both compounds significantly increased to 34382 and 53960 μg g−1 of total HA in fraction 10, thereby confirming the elution behavior noted for TIC data (Table S2 of ESI†). Conversely, the content under the first TIC signal of a highly oxygenated cyclic acid identified with a C7H6O8 empirical formula (Table S2 of ESI†), first increased by passing from size-fraction 1 to 3, and, then, progressively decreased in smaller size-fractions (Table 1).
Although the abundance of individual alkanoic acids, be they saturated, unsaturated, or hydroxyl-unsaturated homologues, did not show any relation to size of eluting fractions (Table 1), a meaningful trend can be found by pooling these into short- and long-chain groups: the short acids up to dodecanoic acid, and the acids longer than tridecanoic acid. In fact, the ratio of short- over long-chain acids did generally increase with decreasing fraction size (Fig. 2), thereby suggesting a prevalence of long chain acids in suprastructures of larger volumes, and an abundance of short chain acids in those of smaller volume. As alkanoic acids were found largely in RES4, their average length may thus be the main factor that controls the size of humic associations in this humeomics end-product. Moreover, the ubiquitous massive presence of alkanoic acids already observed in either humic,6 and fulvic acids,12 as well as in humin,13 further suggests that such a compound class may be mostly responsible for the stability and size of humic supramolecular associations.
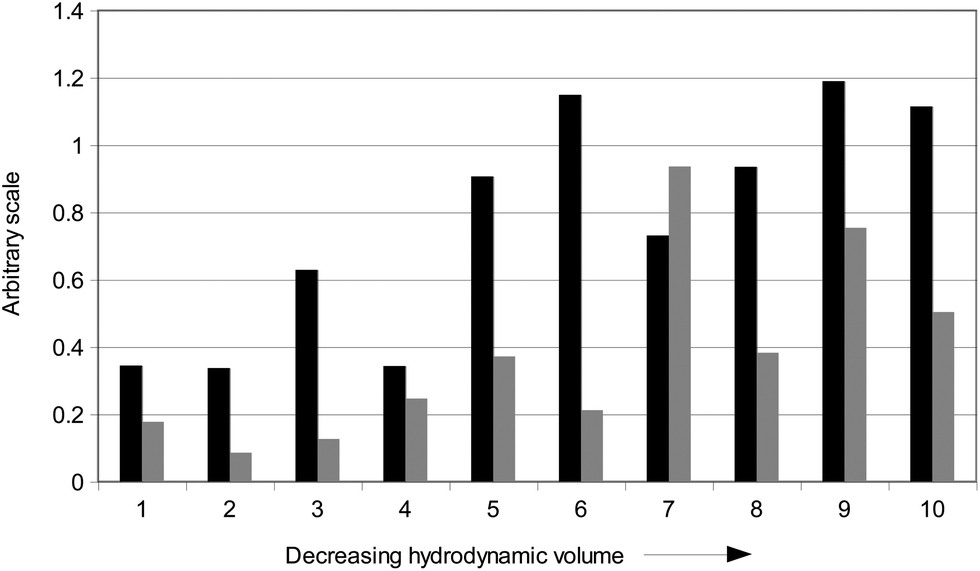 | ||
| Fig. 2 Correlations between elution time of ten size-fractions from RES4 and length (black) and substitution degree (gray) of carbon chain in molecules identified in size-fractions. Values are based on quantitative assessment (Table 1). Black filling: total amount (μg g−1 of total HA weight) of short chain acids divided by total amount (μg g−1 of total HA weight) of long chain acids. Gray filling: total amount (μg g−1 of total HA weight) of both hydroxy unsaturated and unsaturated acids divided by total amount (μg g−1 of total HA weight) of unsubstituted linear alkanoic acids. | ||
By pooling the content of unsaturated and hydroxylated alkanoic acids for each size fraction in one group, and that of unsubstituted alkanoic acids in an another, we attributed a significance to the ratio of the first group over the second one. In fact, the variation of this ratio indicated a non-linear increase with progressive decrease of size-fractions (Fig. 2). As noted earlier, the abundance of unsubstituted acids in larger size-fractions could be attributed to their regular shape and, consequently, more ordered packing in large-sized supramolecular associations. Conversely, alkanoic acids bearing one or more unsaturation or hydroxyl substitution are less sterically able to regularly associate with other humic compounds, and therefore may accumulate preferably in small size-fractions.
A quantitative evaluation of all compounds identified in RES4 and its ten size-fractions, showed that the sum of each analyte in the combined size-fractions significantly exceeded that for the unfractionated bulk RES4 (Table 1). This substantial discrepancy may be to the result of a weaker molecular association in the separated size-fractions than for the bulk material, with consequent easier ionization and detection of molecules in size-fractions. In fact, the original intermolecular association in RES4 was disrupted during HPSEC separation of size-fractions. Reorganization of the separated smaller associations into less stable and less tightly aggregated superstructures facilitated the mass spectrometry analysis by significantly increasing analytical yields. This explanation agrees well with previous findings, which showed that a preliminary size-fractionation of a humic acid enhanced detection yield for molecules undergoing a humeomic procedure.9
We concentrated attention on the four masses with m/z 129, 141, 155 and 217, to which the instrument software attributed the empirical formulae of C6H10O3, C6H6O4, C6H4O5, and C7H6O8, respectively. The mass–mass (MS2) fragmentation spectra for these four empirical formulae are reported in Fig. 3, where the most plausible structure is reconstructed based on the resulting fragmented masses.
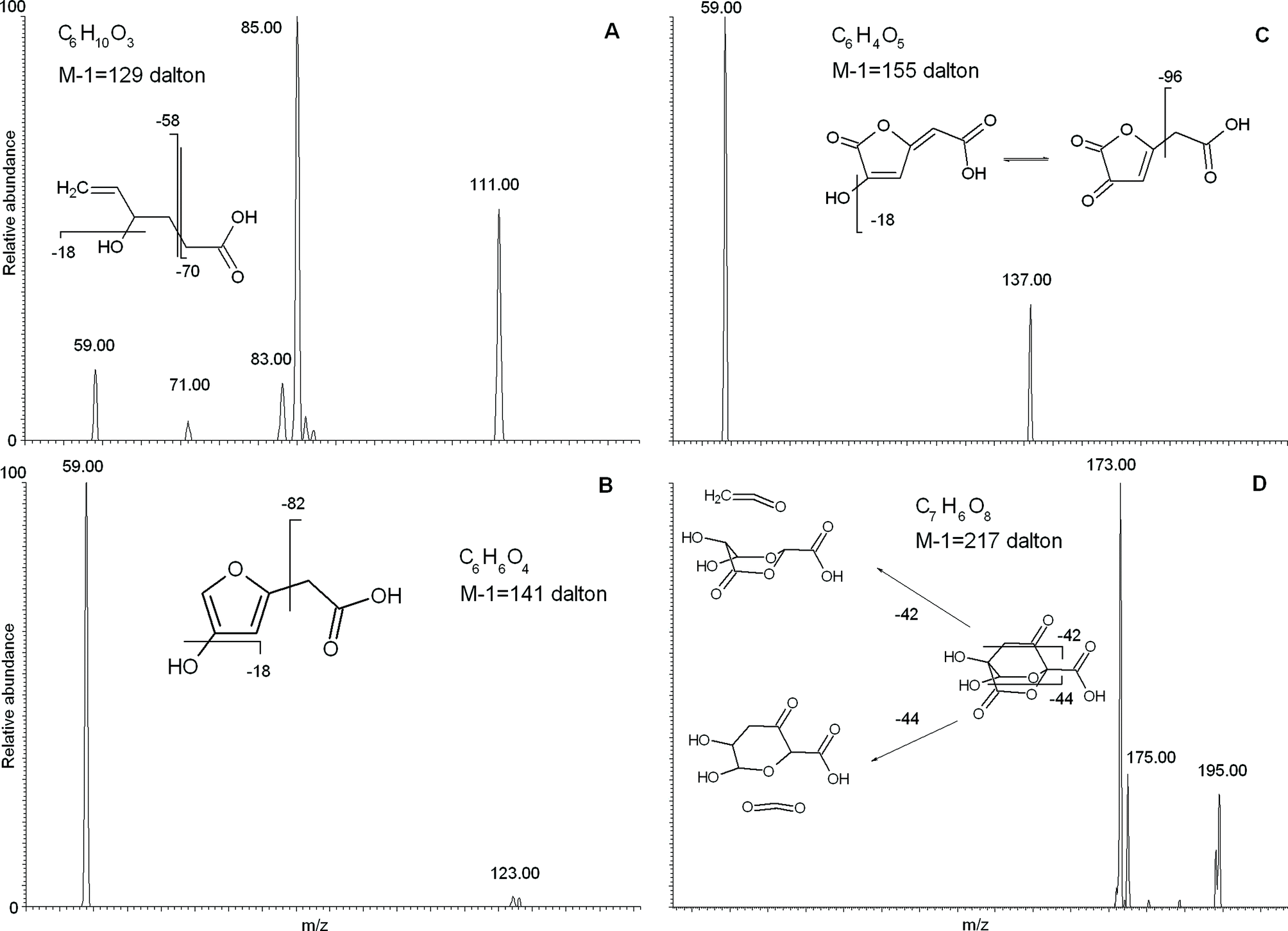 | ||
| Fig. 3 Tandem MS fragmentation analysis and plausible structure interpretation for four compounds with empirical formulae C6H10O3 (a); C6H6O4 (b); C6H4O5 (c); C7H6O8 (d). | ||
The MS2 fragmentation for the C6H10O3 empirical formula (Fig. 3A) showed formation of a [M − 44] daughter ion, which signifies loss of CO2, as commonly occurs for carboxylic acids. Furthermore, the [M − 70] fragment results from a structural breakdown to leave an acetate CH3COO− group, thus confirming the presence of a carboxylic acid. A daughter ion at [M − 18] implies loss of water and, hence, separation of a hydroxyl group. Moreover, the large intensity of the 111 mass peak suggests preferential cleavage of a weak chemical bond between the hydroxyl group and the carbon chain, such as that occurring at a benzylic or allylic position. The identified –CH2COO− and –OH groups accounted for the partial empirical formula of C2H3O3. The remaining atoms represented an empirical formula of C4H6, a butyl chain with a single unsaturation. Further indications were provided by the [M − 58] fragment, which can be attributed to loss of CH2COO− with charge retention on the alkyl chain. In fact, this fragment may be the end-part of a carbon chain bearing a carboxyl group, which, when placed in combination with the unsaturated butyl chain, would produce a hexenoic acid with a single hydroxyl substitution. The fragmented hydroxyl group should be placed on the C4 with respect to the carboxyl group, as the C5 or C6 positions must be part of a double bond, because of the unlikeliness of an unstable tautomer such as a vinyl alcohol. The adjacent C3 position was also ruled out for hydroxylation, because this would favor a great intensity for the [M − 41] ion as a stable fragment of hydroxypropanoic acid, which was absent in the fragmentation pattern. Thus, the allylic position for the hydroxyl group is most likely, as this would explain the relatively strong intensity of the daughter ion caused from loss of water. Thus, the structure of the compound with a C6H10O3 empirical formula mostly consistent with experimental data is that reported in Fig. 3A.
The molecule with a m/z 141 mass and C6H6O4 as empirical formula was also characterized by MS2 fragmentation (Fig. 3B). The precursor compound produced [59] and [M − 18] as main daughter ions, which were interpreted as occurring with formation of acetic acid and neutral loss of water, respectively. With the same logic used above (Fig. 3A), these daughter ions may be explained with a hydroxyl group and a carboxyl group with an unsubstituted alpha position. Then, by subtracting an acetic group and a hydroxyl group from the original empirical formula, the residual C4H3O may be explained with an unsaturated furane ring. Arrangement of the identified hydroxyl and acetic groups on the furane ring yielded the structure shown in Fig. 3B. This structure accounts for the lower intensity of the [M − 18] daughter ion, as the energy required to break the OH–furane bond and release water is greater than that for the hydroxyhexenoic acid (Fig. 3A).
Similarly, the [59] and [M − 18] daughter ions obtained by MS2 fragmentation of the empirical formula related to the m/z 155 mass peak, were attributed to acetic acid and neutral loss of water, respectively (Fig. 3C). The C4H1O2 empirical formula remaining after subtraction of acetic acid and water from the initial C6H4O5, may be justified by an oxidized oxo-furane ring (Fig. 3C). Although it could be argued that loss of water from an oxo-substituent is unlikely, it is true that its tautomeric form is capable of such fragmentation (Fig. 3C). The energy required to break such a vinyl OH from carbon chain would be significant, and this explains why the intensity of its consequent [M − 18] daughter ion was relatively lower than for the corresponding fragmentation in hydroxyhexenoic acid (Fig. 3A). The furane structures with empirical formulae with m/z 141 and 155 may also suggest a possible carbohydrate source. In fact, extensive oxidation of a carbohydrate structure may produce a carboxyl group and condensation into a furane ring.
Finally, for the empirical formula C7H6O8 with m/z 217 (Fig. 3D), the MS2 fragmentation produced strong peaks with m/z [M − 42] and [M − 44] and a weaker one with m/z [M − 18], attributable to loss of C2H2O (ketene), CO2, and H2O groups, respectively. A loss of a saturated C3H7 propyl group was ruled out as an alternative explanation for the [M − 42] daughter ion, because of a number of H atoms even greater than the parent C7H6O8 compound. Conversely, loss of ketene was a plausible reason for the formation of [M − 42], as this fragmentation already had been reported for other cyclic terpene molecules, such as esters and ketones with endocyclic –CH2–CO–.14 The subtraction of ketene, CO2, and water from the parental molecule produced the residual C4H2O4 empirical formula, which could not be accounted for by a furane ring because of instability of endocyclic structures with –CH2–CO– groups. A 1,4 dioxane ring with two oxygen substituents proved a better candidate to provide endocyclic substitution, such as the compound suggested in Fig. 3D. In such a structure, a –CH2–CO– chain binds together the C2 and C5 carbons in a norbornane-like ring (Fig. 3D), and bears both a carboxyl and a hydroxyl group as additional substituents. This structure fits with the experimental data and satisfies both its empirical formula and unsaturation index. Furthermore, it may explain the unusually intense [M − 44] fragmentation peak, as the related CO2 loss may occur from the breakdown of endocyclic ester, by the same mechanism as discussed above for the ketene loss from an endocyclic ketone (Fig. 3D). The structural complexity of this molecule is unusual when compared with the abundance of linear compounds found in the bulk RES4 and its size fractions. The possible origin of such a norbornane-type structure is decay of secondary metabolites released by plants.
It is noteworthy that the norbornane-type structure proposed here to explain a mass contained in the recalcitrant RES4 end-product, may well be related to the so-called carboxyl-rich alicyclic molecules (CRAM). This class of compounds is defined as fused carboxylated alicyclic structures, with a carboxyl-C/aliphatic-C ratio between 1:2 and 1:7, and generally have been reported in dissolved organic matter (DOM) by NMR and ultra high resolution mass spectrometry.9,15−17 Although CRAM are generally regarded as refractory components of dissolved organic matter in marine environments,16 the presence in NOM of terrestrial origin with a structure with great similarity to CRAM is reported here for the first time. Thus, this finding suggests that cyclic acids and CRAM are formed by similar biogeochemical pathways, and their different environmental fates ultimately may be determined by their degree of hydrophobicity.
3. Experimental section
3.1 Humeomic fractionation
All reagents were from Sigma-Aldrich (99.9% pure), and were used without further purification. A humic acid (HA) was isolated from volcanic soil (Allic Fulvudand) sampled at Vico lake, near Rome (Italy), and purified as described elsewhere.18 This HA was then submitted to the step-wise humeomics procedure as previously reported in detail.6,9 The procedure removed either unbound or ester-bound molecules from humic matrix, as well as ether-bound molecules, and left a final solid residue defined as RES4. This was extensively washed with Milli-Q deionized water until the disappearance of iodine, and freeze-dried. The freeze-dried solid RES4 humic residue was suspended in water and dissolved with a 0.50 M NaOH solution by automatically titrating under N2 to pH 7.2 (VIT 909 21 Videotitrator, Radiometer, Copenhagen) until pH remained constant for 120 min, reaching a final concentration of 0.20 g L−1. Possible microbial growth was prevented by adding 0.3 g L−1 NaN3. This RES4 solution was then filtered through a 1 μm glass microfiber filter (Whatman GF/C) and kept refrigerated under N2 atmosphere, until being subjected to preparative high performance size exclusion chromatography (HPSEC).3.2 Preparative HPSEC
The RES4 solution was eluted through a Phenomenex Biosep SEC-S-2000 column (21.2 mm diameter × 300 mm length) and precolumn (21.2 mm diameter × 78 mm length). A Gilson 305 pump, a Gilson auto-sampler model 231 equipped with a 5.0 mL loop, a Gilson FC205 fraction collector, and a Gilson 116 UV detector set at 280 nm were used to automatically and continuously generate humic fractions. Chromatographic runs and profiles were monitored with a Gilson Unipoint software. A mobile phase consisting of aqueous (Milli-Q Millipore deionized water) 0.3 g L−1 NaN3, 0.01 M AcONa, and 3.5 mM of NaH2PO4/Na2HPO4 buffer solution at pH 7.0 was eluted through columns at a flow of 1.5 mL min−1. A total of 50 mg of RES4 was injected into the preparative system and ten fractions were separated at different elution times. Yields for the ten fractions are summarized in Table S1 of ESI.† The 10 fractions (F1–F10) were acidified to pH 2.0 with 1.0 M HCl, and the precipitated humic matter was dialyzed against Milli-Q deionized water in Spectrapore 3 membranes (cut-off 3500 Da), and freeze-dried.3.3 Hyphenated HPSEC-ESI-MS analysis
Solutions (0.4 g L−1) of the original RES4 and its ten size separates were made in 0.01 M NH3, transferred into LC vials, and injected into a HPSEC-MS system by a 50 μL Rheodyne loop. The flow was generated by a Dionex P 580 pump working at 0.3 mL min−1 elution rate. The mobile phase was a mixture of two solutions A/B in a ratio of 55/45 (A: 5 mM AcONH4 in Milli-Q water and 5% MeCN, pH 7; B: 100% MeCN) and was pumped through a Phenomenex Bio-Sep SEC-S 2000 analytical column (300 × 7.8 mm) and precolumn (30 × 7.8 mm), both thermostatted at 30 °C. This HPSEC system was directly connected to a LTQ Orbitrap (Thermo Electron, Waltham, MA) mass-spectrometer, which acquired spectra with negative ESI mode, a mass range of 100–1000 m/z, and a 1.0 s scan time. N2 was used as the sheath gas (45 AU) and He as collision gas (7.99 AU). Spray voltage was set at 4.00 kV, spray current at 2.05 μA, capillary temperature at 260 °C, and capillary voltage at 14.93 V. Tandem MS experiments were conducted in LTQ mode at a resolution of 7500 m/z. For each compound, a scan window ranging from 50 m/z to the mass of the molecular ion was programmed. A collision energy of 45 units for MS2 and 35 for MS3 was applied. Average scan time was set at 30 ms for each scan event.External standard solutions were prepared with isotopically labeled compounds (Cambridge Isotope Labs, 99%), such as ω-deuterated hexadecanoic acid (16-d-3) for linear compounds and 13C labeled 4-OH-benzoic acid (ring 13C-6) for cyclic compounds. A stock solution of 1.0 g L−1 in 0.01 M NH3 was prepared for each standard and subsequently diluted to reach a concentration of 10 mg L−1. Standards were obtained by diluting this stock solution at the following concentrations: 10 and 100 μg L−1, and 1 and 10 mg L−1. Additionally, a blank was prepared without standard. A calibration curve covering variations by five orders of magnitude was built with instrumental response on the basis of standards and blank.
Internal standard solutions were prepared by first dissolving aliquots of RES4 and its size-fraction in 0.01 M NH3 to reach a 0.40 g L−1 final concentration. After centrifugation, the resulting supernatant was spiked with the previously described stock solution of isotopically labeled compounds (1.0 g L−1), to reach the same concentration in humic solutions as the external standards: blank (no standard), 10 and 100 μg L−1, and 1 and 10 mg L−1. A calibration curve was built according to instrumental responses of internal standards.
4. Conclusions
Our results showed that even a recalcitrant end-product (RES4) of humeomics applied to a soil humic acid could be successfully solubilized in alkaline solution, and its heterogeneity reduced by separating ten different size-fractions by preparative HPSEC. Further decrease of the complex intermolecular humic associations was obtained by eluting the size-fractions through an analytical HPSEC column before characterization by a hyphenated a high-resolution Orbitrap ESI-MS. We found that the molecular components of size-fractions consisted of alkanoic acids, unsaturated alkanoic and hydroxyalkanoic acids, other linear unsaturated oxygenated acids, and oxygenated cyclic acids. Moreover, application of tandem high-resolution mass spectrometry to masses of uncertain structure allowed their most plausible identification as unsaturated hydroxy carboxylic acids, furane rings, and complex norbornane-like compounds. The molecular elucidation of such highly unsaturated and oxidized chemical structures is unprecedented for terrestrial humic matter, and suggests that extensive application of tandem mass spectrometry may help to enhance our knowledge on the elusive quaternary-carbon components of humic matter. For so long, a lack of characterization of recalcitrant humic matter has prevented progress in molecular understanding of natural organic matter; however, we confirm here that humeomics can overcome this limitation and promises to further enhance knowledge on the structural composition of natural organic matter and its dynamics in environmental compartments.Acknowledgements
The first author is grateful for the collaboration provided by the Institut für Umweltforschung of the Technical University of Dortmund.References
- F. J. Stevenson, Humus chemistry: genesis, composition, and reactions, Wiley-Interscience, New York, 2nd edn, 1994 Search PubMed.
- A. Piccolo, Humus and soil conservation, in Humic Substances in Terrestrial Ecosystems, ed. A. Piccolo, Elsevier, Amsterdam, Netherlands, 1996, pp. 225–264 Search PubMed.
- S. Nardi, D. Pizzeghello, A. Muscolo and A. Vianello, Physiological effects of humic substances on higher plants, Soil Biol. Biochem., 2002, 34, 1527–1536 CrossRef CAS.
- A. Piccolo, The supramolecular structure of humic substances: A novel understanding of humus chemistry and implications in soil science, Adv. Agron., 2002, 75, 57–134 CAS.
- D. Šmejkalová and A. Piccolo, Aggregation and disaggregation of humic supramolecular assemblies by NMR diffusion ordered spectroscopy (DOSY-NMR), Environ. Sci. Technol., 2008, 42, 699–706 CrossRef.
- A. Nebbioso and A. Piccolo, Basis of a humeomics science: chemical fractionation and molecular characterization of humic biosuprastructures, Biomacromolecules, 2011, 12, 1187–1199 CrossRef CAS PubMed.
- A. Piccolo, P. Conte, E. Trivellone, B. Van Lagen and P. Buurman, Reduced heterogeneity of a lignite humic acid by preparative HPSEC following interaction with an organic acid. Characterization of size-separates by Pyr-GC-MS and 1H-NMR spectroscopy, Environ. Sci. Technol., 2002, 36, 76–84 CrossRef CAS.
- P. Conte, R. Spaccini and A. Piccolo, Advanced CPMAS-13C NMR techniques for molecular characterization of size-separated fractions from a soil humic acid, Anal. Bioanal. Chem., 2006, 386, 382–390 CrossRef CAS PubMed.
- A. Nebbioso and A. Piccolo, Advances in humeomics: Enhanced structural identification of humic molecules after size fractionation of a soil humic acid, Anal. Chim. Acta, 2012, 720, 77–90 CrossRef CAS PubMed.
- K. Matsuda and M. Schnitzer, The permanganate oxidation of humic acids extracted from acid soils, Soil Sci., 1972, 114, 185–193 CrossRef CAS PubMed.
- O. B. Maximov, T. V. Shvets and Y. N. Elkin, On permanganate oxidation of humic acids, Geoderma, 1977, 19, 63–78 CrossRef CAS.
- M. Schnitzer and J. A. Neyroud, Alkanes and fatty acids in humic substances, Fuel, 1975, 54, 17–19 CrossRef CAS.
- A. Nebbioso and A. Piccolo, The molecular composition of soil Humin is unravelled by Humeomics, Soil Biol. Biochem., 2014 Search PubMed , under review.
- T. Donovan and J. Brodbelt, Characterization of the dissociation behavior of gas-phase protonated and methylated lactones, J. Am. Soc. Mass Spectrom., 1992, 3, 47–59 CrossRef CAS.
- M. V. McCaul, D. Sutton, A. J. Simpson, A. Spence, D. J. McNally, B. W. Moran, A. Goel, B. O'Connor, K. Hart and B. P. Kelleher, Composition of dissolved organic matter within a lacustrine environment, Environ. Chem., 2011, 8, 146–154 CrossRef CAS.
- N. Hertkorn, R. Benner, M. Frommberger, P. Schmitt-Kopplin, M. Witt, K. Kaiser, A. Kettrup and J. I. Hedges, Characterization of a major refractory component of marine dissolved organic matter, Geochim. Cosmochim. Acta, 2006, 70, 2990–3010 CrossRef CAS PubMed.
- N. Hertkorn, M. Harir, B. P. Koch, B. Michalke and P. Schmitt-Kopplin, High-field NMR spectroscopy and FTICR mass spectrometry: powerful discovery tools for the molecular level characterization of marine dissolved organic matter, Biogeosciences, 2013, 10, 1583–1624 CAS.
- A. Nebbioso and A. Piccolo, Molecular rigidity and diffusivity of Al3+ and Ca2+ humates as revealed by NMR spectroscopy, Environ. Sci. Technol., 2009, 43, 2417–2424 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra01619j |
| This journal is © The Royal Society of Chemistry 2014 |