
Nitrogen/sulfur co-doped non-noble metal material as an efficient electrocatalyst for the oxygen reduction reaction in alkaline media
Abstract
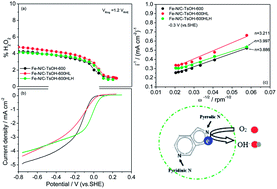
This work demonstrates the feasibility of nitrogen/sulfur co-doped non-noble metal materials (Fe–N/C–TsOH) as platinum-free catalysts for the oxygen reduction reaction (ORR) in alkaline media. Electrochemical techniques such as cyclic voltammetry (CV), rotating disk electrodes (RDEs) and rotating ring-disk electrodes (RRDEs) are employed with the Koutecky–Levich theory to investigate the ORR kinetic constants and the reaction mechanism. It is found that the catalysts doped with TsOH (p-toluenesulfonic acid) show significantly improved ORR activity relative to a TsOH-free catalyst. The overall electron transfer numbers for the catalyzed ORR are determined to be 3.899 and 3.098, respectively, for the catalysts with and without TsOH-doping. Catalysts heat treated at 600 °C exhibit relatively higher activity. In addition, the catalyst doped with TsOH (Fe–N/C–TsOH-600) not only exhibits exceptional stability in 0.1 mol L−1 KOH solution but also has higher methanol tolerance compared to commercial Pt/C catalyst in 0.1 mol L−1 KOH. To some extent, increasing the Fe–N/C–TsOH-600 loading on the electrode favors a faster reduction of H2O2 to intermediate to H2O. X-ray photoelectron spectroscopy analysis indicates that pyrrolic N groups are the most active sites, and that sulfur species are structurally bound to carbon in the forms of C–S(n)–C and oxidized –SO(n)– bonds, an additional beneficial factor for the ORR.
 Please wait while we load your content...
Please wait while we load your content...

