
A rapid and modified approach for C-7 amination and amidation of 4-methyl-7-nonafluorobutylsulfonyloxy coumarins under microwave irradiation†
Abstract
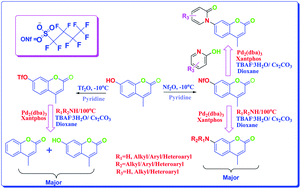
A facile, efficient and reliable access for the synthesis of an array of 4-methyl-7-(alkyl/aryl/heteroaryl) amino and amido coumarins has been developed by treating 4-methyl-7-nonafluorobutylsulfonyloxy coumarin with various amines and pyridones in presence of a catalyst combination of Pd2(dba)3/xantphos under microwave irradiation. Nonaflates coupled efficiently to give the diaryl amines in acceptable to excellent yields whereas the corresponding triflate was unstable, yielding the detriflated product as well as the hydrolyzed product as competing side products along with the desired product. The wide bite angle (108°) of xantphos, employment of Cs2CO3 as a mild base and the utilization of TBAF·3H2O as an additive proved to be key for success of the reaction.
 Please wait while we load your content...
Please wait while we load your content...

