Anti-HSV activity and mode of action study of α-pyrone carboxamides†
Srinivas Karampuri‡
a,
Durbadal Ojha‡b,
Paromita Bagb,
Harapriya Chakravartya,
Chandralata Bala,
Debprasad Chattopadhyay*b and
Ashoke Sharon*a
aDepartment of Applied Chemistry, Birla Institute of Technology, Mesra, Ranchi 835215, India. E-mail: asharon@bitmesra.ac.in; Fax: +91-651-2275401; Tel: +91-651-2276531
bICMR Virus Unit, ID & BG Hospital, Beliaghata, Kolkata 700010, India. E-mail: debprasadc@yahoo.co.in; Fax: +91-33-2353-7424; Tel: +91-33-2353-7425
First published on 20th February 2014
Abstract
The clinical management of herpes virus diseases is limited due to ineffective clearance of virus particles and frequent emergence of drug-resistant viruses, particularly in immunocompromised patients, pregnant women and neonates. In our continued quest for new antiviral leads, α-pyrone carboxamide propanol derivatives were synthesized and evaluated in HSV infected Vero cells. Compound 3d showed potent antiviral activity against HSV-IF (EC50 = 9.8 μg ml−1 and EC99 = 18.0 μg ml−1) and HSV-2G (EC50 = 12.4 μg ml−1 and EC99 = 24.0 μg ml−1) at 4–6 h post-infection. The mode of action studies demonstrated that 3d did not interfere in viral attachment or penetration, however, it reduced the expression of ICP4 and ICP27 (immediate-early gene products) as well as the HSV DNA polymerase.
1. Introduction
The Herpesviridae are a large family of DNA viruses that include Herpes Simplex Virus type 1 and type 2 (HSV-1 and HSV-2). HSV-1 and HSV-2 are responsible for the cold sores and genital herpes that have plagued humanity for centuries. DNA polymerase is one of the most important early proteins that play a key role during replication of these viruses. HSV and human DNA polymerase belong to the family of type B DNA polymerases and share six to seven highly conserved domains including domain C.1 We are focused on discovering new non-nucleoside analogs (NNA) capable of selective inhibition of viral DNA polymerase. This may lead to the achievement of desirable inhibition with lesser side effects and toxicity. PNU-183792 (I, Fig. 1) having broad spectrum anti-HSV activity with high specificity for viral polymerases compared to human polymerases, is a 4-oxo-dihydroquinoline-3-carboxamide (DHQ) based derivative.2 An earlier report shown that DHQ molecules were active in plaque reduction assays against various isolates of HSV and the average IC50s were found in the range of 14 ± 4.0 μM, in comparison to reference drug acyclovir (ACV) of 8.6 ± 5.4 μM.2 Further, studies on promising analog I have shown comparable drug-like properties with respect to licensed drugs acyclovir, ganciclovir, and cidofovir.3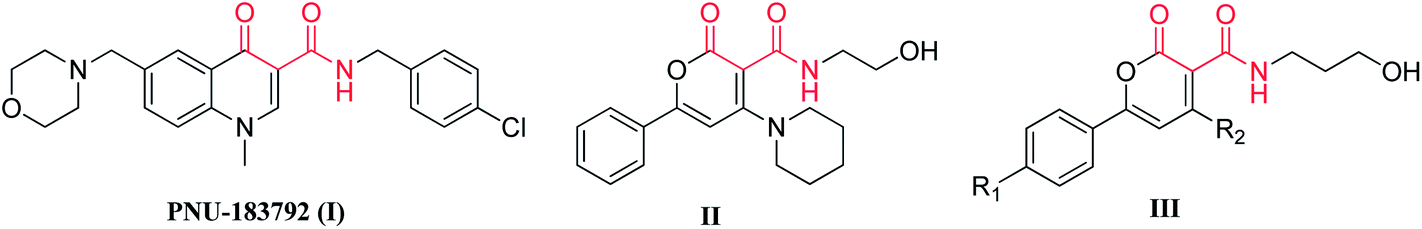 | ||
| Fig. 1 Chemical structure of potential compound PNU-183792 (I), preliminary lead (II) and new prototype molecules (III). | ||
ACV, a synthetic guanine nucleoside analog, is a prodrug that is phosphorylated to ACV monophosphate preferentially by thymidine kinase of the herpes virus, which is then further phosphorylated to the di and tri-phosphate forms by cellular enzymes. The pharmacologically active ACV-triphosphate then inhibits the viral DNA polymerase along with chain termination. Despite the availability of ACV and its analogues, HSV can cause life-threatening infections such as herpes encephalitis. Neither acyclovir nor its analogues can eliminate the virus in the latent state;4 while their extensive clinical use leads to the emergence of drug-resistant viruses.5 Consequently, continuation of the HSV pandemic is anticipated and efforts to develop vaccines and novel therapies aimed at eliminating the latent virus are still warranted.6,7 However, a recent study showed that anti-HSV therapy significantly reduces HIV-1 RNA load in co-infected patients.8 Therefore, alternative treatments to minimize the development of resistance, reduce side-effects with better efficacy are urgently needed. To date no antiviral compound has been able to completely inhibit the replication of any virus and a proportion of viral particles always seem to be able to circumvent the drug-induced blockade.
In our recent report, we described the inclusion of the pharmacophoric motif (red) of I for designing new molecules based on α-pyrone carboxamide, and identified II (Fig. 1) as our preliminary lead candidate with EC50 of 12.4 μg ml−1 and CC50 of 52.4 μg ml−1.9 The identification of the pharmacophoric motif was further followed by modeling studies and the preliminary lead compound (II) showed the possibility to fit into the allosteric site in the palm region of HSV DNA polymerase. In addition, the two cavities were identified in the allosteric active site of polymerase, which were utilizing by piperidine and alcoholic motifs of II. Thus, to explore the possibility of our hypothesis, we extended our synthesis in this direction, so that target compounds can utilize these cavities more significantly to enhance anti-HSV activity. Therefore, new analogs (III, Fig. 1) were selected with one more carbon (alcoholic motif) for synthesis, which may allow the molecule to utilize the remaining space of the identified cavity to enhance anti-HSV activity. We have also carried out the modeling studies of III to increase the understanding of the molecular basis of compound binding in the allosteric site of viral DNA polymerase. If the allosteric binding phenomenon exists, it opens a scope to evaluate the synergistic role of this compound (nonnucleoside inhibitor) with nucleoside inhibitor acyclovir. Thus, mode of action studies including possible synergism were also carried out for one of the molecules with licensed drug ACV to test our hypothesis.
2. Results and discussion
2.1 Synthesis of α-pyrone carboxamide derivatives
The α-pyrone-3-carboxylic acid derivatives (1a–c) were synthesized from appropriate acetophenone derivatives (Scheme 1).9 The carboxyl group of 1a–c was coupled with propanolamine using HATU as the coupling reagent to generate the corresponding amides (2a–c). The methylthio group at C-4 of 2a–c was replaced with secondary amines by heating in dioxane to yield 3a–i. | ||
| Scheme 1 Reagents and conditions: (i) propanolamine, HATU, DMF, rt, 6 h; (ii) appropriate amine, dioxane, 70 °C, 4 h. | ||
All the intermediates and final compounds were adequately characterized by spectroscopic analysis, and one of the potential lead compounds 3d was further confirmed by a single crystal X-ray analysis.10
The ellipsoidal plot as an ORTEP diagram is shown with 30% probability in Fig. 2. The piperidine motif was found disordered, seen by two position of the N7 atom at N7A and N7B, and it may be associated to conformational flipping in the chair conformation of the piperidine ring. The complete X-ray structure was solved and refined with good resolution.
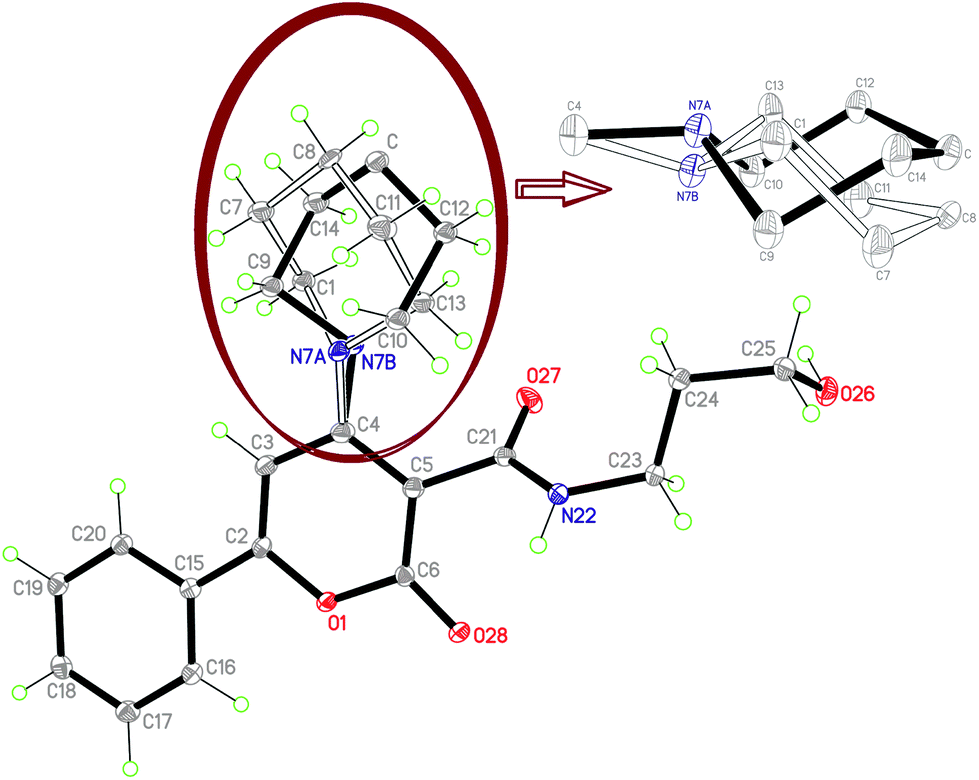 | ||
| Fig. 2 ORTEP diagram of 3d showing the X-ray molecular structure at 30% probability level. The piperidine motif at C4 was found disordered due to conformational flipping in the chair form of piperidine in the crystal structure. | ||
2.2 Biological evaluation
| Compound | CC50a (μg ml−1) | EC50b (μg ml−1) | SIc (CC50/EC50) | EC99 (μg ml−1) | |||
|---|---|---|---|---|---|---|---|
| HSV-1F | HSV-2G | HSV-1F | HSV-2G | HSV-1F | HSV-2G | ||
| a The 50% cytotoxic concentration for Vero cells (CCL 81) in μg ml−1.b Concentration of compound (μg ml−1) producing 50% inhibition of virus induced CPE of three separate experiments.c Selectivity index (SI) = CC50/EC50 [SI index below 4 is insignificant]. ND, Not detected, as the anti-HSV activity was either equal to or above the CC50 concentrations. The EC99 was above the CC50 concentrations for most of the compounds. | |||||||
| II | 52.4 | 12.4 | 13.1 | 4.23 | 4.0 | 22 | 28 |
| 3a | 92.5 | 37.2 | 46.2 | 2.49 | 2.0 | 40 | 42 |
| 3b | 39.4 | 36.4 | >39 | 1.08 | ND | >39 | >39 |
| 3c | 98.2 | 95 | >98 | 1.03 | ND | >98 | >98 |
| 3d | 151.4 | 9.8 | 12.4 | 15.4 | 12.2 | 18 | 24 |
| 3e | 29.2 | 12 | 15 | 2.43 | 1.94 | 20 | 25 |
| 3f | 43.6 | 40 | >43 | 1.09 | ND | >40 | >40 |
| 3g | 76.8 | 33.1 | 72 | 2.32 | 1.07 | 40 | >72 |
| 3h | 117.5 | 112.5 | >117 | 1.04 | ND | 115 | >117 |
| 3i | 37.8 | 36.5 | >37 | 1.03 | ND | >37 | >37 |
| Acyclovir | 136.8 | 2.4 | 2.8 | 57.0 | 48.8 | 5.0 | 6.2 |
 | ||
| Fig. 3 Dose dependent effects of 3d and acyclovir against HSV-1F and HSV-2G. No inhibition was noticed with 0.1% DMSO, used as solvent (data not shown). | ||
 | ||
| Fig. 4 Time of addition assay of 3d and acyclovir against HSV-1F. | ||
 | ||
| Fig. 5 Effects of 3d and acyclovir on HSV-1F attachment and penetration. | ||
| Compound | EC50 | FICcompound + FICACV | Inhibitory effect |
|---|---|---|---|
| HSV-1F | HSV-1F | ||
| a The interaction between 3d and ACV was interpreted according to the combined FIC index [FICcompound + FICACV] as synergy (≤0.5), no interaction (0.5–4) or antagonism (>4) (32). | |||
| 3d alone | 9.8 ± 0.1 | — | — |
| Acyclovir alone | 2.4 ± 0.1 | — | — |
| Acyclovir + 3d | 0.75 ± 0.1 | 0.75/9.8 + 0.75/2.4 = 0.389 | Synergism |
 | ||
| Fig. 6 Effect of 3d on IE gene expression of HSV-1F by quantitative real time PCR. | ||
 | ||
| Fig. 7 Effect of 3d on IE gene expression by Western blot analysis. | ||
2.3 Molecular modeling studies
In our previous report, we suggested that the binding cleft for pyranone class of molecules may be positioned in the palm sub-domain of HSV DNA polymerase (Fig. 8a).3 When the preliminary lead compound (II) was docked into the active site, cavity A was partially occupied by the ethanol motif and cavity B was fully occupied by the piperidine motif (Fig. 8b). | ||
| Fig. 8 (a) Binding cleft for pyranone class of molecules (shown as black surface) in the palm region of HSV DNA polymerase active site. (b) Surface diagram for binding of II into the palm region using cavities A and B. (c) The surface diagram showing the electrostatic surface of the receptor for the binding of 3d into the palm region using cavities A and B. | ||
Thus in the present manuscript, cavity A was explored by prototypes III in which the ethanol motif was replaced with a propanol motif. The surface diagram for one of the prototypes (3d, Fig. 8c) reveals a similar binding mode as II inside the palm region. However, the propanol motif occupied more space in cavity A. The binding scores of II and prototypes III summarized in Table 3 indicate an increase in overall binding affinity of 3d compared to II which may be the possible reason for the enhanced activity. Thus, the present study supports lead optimization to enhance the anti-HSV activity using a structure-based approach.
| Docking | MM-GBSA scorec | ||
|---|---|---|---|
| Compound | G-Scorea | Emodelb | DG bind |
| a G-Score (Glide Score): the minimized poses are rescored using Schrödinger's proprietary Glide Score scoring function to get G-Score.b Emodel score: the binding affinity can be estimated by Glide Emodel Score, which is the combination of G-Score and non-bonded interaction energy.c MM-GBSA score: ligand binding energies calculated for all compounds with a single receptor using protein flexibility option (defined: 7 Å from docked ligand). Solvation model: VSGB, The binding energy is calculated according to the equation: DG bind = Ecomplex (minimized) − Eligand (minimized) − Ereceptor (minimized). | |||
| 3a | −4.8 | −64.9 | −59.9 |
| 3b | −6.1 | −63.8 | −68.4 |
| 3c | −5.6 | −62.1 | −69.5 |
| 3d | −5.7 | −67.1 | −72.3 |
| 3e | −5.6 | −69.1 | −73.1 |
| 3f | −5.1 | −61.6 | −76.8 |
| 3g | −5.2 | −60.7 | −62.3 |
| 3h | −5.6 | −65.4 | −60.7 |
| 3i | −5.1 | −63.6 | −60.7 |
| II | −4.9 | −64.9 | −68.7 |
3. Materials and methods
3.1 Chemistry
N-(3-Hydroxypropyl)-2-oxo-6-phenyl-4-(pyrrolidin-1-yl)-2H-pyran-3-carboxamide (3a). Yield 55%, mp: 144–145 °C, MS-ESI (m/z): 342.9 [M+ + 1]; IR (KBr) (ν, cm−1): 3360.00 (NH), 3290 (OH), 1655 and 1624 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (400 MHz, DMSO-d6): δ 1.59–1.62 (t, J = 6.8 Hz, 2H), 1.88 (s, 4H), 3.18–3.21 (t, J = 6.8 Hz, 2H), 3.44–3.47 (t, J = 6 Hz, 2H), 3.53 (s, 4H), 4.41–4.44 (t, J = 5.2 Hz, 1H), 6.79 (s, 1H), 7.51–7.53 (m, 3H),7.89–7.90 (m, 2H), 8.06–8.09 (t, J = 5.6 Hz, 1H).
O); 1H NMR (400 MHz, DMSO-d6): δ 1.59–1.62 (t, J = 6.8 Hz, 2H), 1.88 (s, 4H), 3.18–3.21 (t, J = 6.8 Hz, 2H), 3.44–3.47 (t, J = 6 Hz, 2H), 3.53 (s, 4H), 4.41–4.44 (t, J = 5.2 Hz, 1H), 6.79 (s, 1H), 7.51–7.53 (m, 3H),7.89–7.90 (m, 2H), 8.06–8.09 (t, J = 5.6 Hz, 1H).
6-(4-Fluorophenyl)-N-(3-hydroxypropyl)-2-oxo-4-(pyrrolidin-1-yl)-2H-pyran-3-carboxamide (3b). Yield 52%, mp: 188–189 °C, MS-ESI (m/z): 360.9 [M+ + 1]; IR (KBr) (ν, cm−1): 3365 (NH), 3333 (OH), 1670 and 1639 (C
O); 1H NMR (400 MHz, DMSO-d6): δ 1.59–1.62 (t, J = 6.8 Hz, 2H), 1.88 (brs, 2H), 3.19–3.22 (t, J = 6.4 Hz, 2H), 3.43–3.48 (q, J = 6 Hz, 2H), 3.52 (brs, 4H), 4.41–4.44 (t, J = 5.2 Hz, 1H, OH), 6.78 (s, 1H),7.34–7.38 (t, J = 8.8 Hz, 2H),7.95–7.99 (m, 2H), 8.05–8.08 (t, J = 6 Hz, 1H, NH).
N-(3-Hydroxypropyl)-2-oxo-4-(pyrrolidin-1-yl)-6-(p-tolyl)-2H-pyran-3-carboxamide (3c). Yield 55%, mp: 135–136 °C, MS-ESI (m/z): 357.1 [M+ + 1]; IR (KBr) (ν, cm−1): 3362 (NH), 3298 (OH), 1661 and 1628 (C
O); 1H NMR (400 MHz, DMSO-d6): δ 1.59–1.62 (t, J = 6.4 Hz, 2H), 1.87 (s, 4H), 2.36 (s, 3H), 3.17–3.21 (t, J = 6.4 Hz, 2H), 3.45–3.48 (t, J = 6 Hz, 2H), 3.52 (s, 4H), 4.40–4.42 (t, J = 5.6 Hz, 1H, OH), 6.73 (s, 1H), 7.31–7.33 (d, J = 8 Hz, 2H), 7.78–7.80 (d, J = 8.4 Hz, 2H), 8.02–8.05 (t, J = 5.6 Hz, 1H, NH).
N-(3-Hydroxypropyl)-2-oxo-6-phenyl-4-(piperidin-1-yl)-2H-pyran-3-carboxamide (3d). Yield 59%, mp: 172–173 °C, MS-ESI (m/z): 356.9 [M+ + 1]; IR (KBr) (ν, cm−1): 3362 (NH), 3289 (OH), 1656 and 1627 (C
O); 1H NMR (400 MHz, DMSO-d6): δ 1.59–1.62 (m, 8H), 3.18–3.23 (t, J = 6.8 Hz, 2H), 3.39–3.46 (m, 6H), 4.42–4.45 (t, J = 5.2 Hz, 1H, OH), 6.98 (s, 1H), 7.51–7.52 (t, J = 3.2 Hz, 3H), 7.91–7.92 (d, J = 2.8 Hz, 2H), 8.15 (s, 1H, NH).
6-(4-Fluorophenyl)-N-(3-hydroxypropyl)-2-oxo-4-(piperidin-1-yl)-2H-pyran-3-carboxamide (3e). Yield 54%, mp: 166–167 °C, MS-ESI (m/z): 374.9 [M+ + 1]; IR (KBr) (ν, cm−1): 3367 (NH), 3306 (OH), 1666 and 1628 (C
O); 1H NMR (400 MHz, DMSO-d6): δ 1.59–1.62 (m, 8H), 3.18–3.23 (q, J = 6.8 Hz, 2H), 3.43–3.47 (m, 6H), 4.42–4.45 (t, J = 5.2 Hz, 1H, OH), 6.97 (s, 1H), 7.34–7.38 (t, J = 8.8 Hz, 2H), 7.98–8.02 (m, 2H), 8.13–8.16 (t, J = 5.6 Hz, 1H, NH).
N-(3-Hydroxypropyl)-2-oxo-4-(piperidin-1-yl)-6-(p-tolyl)-2H-pyran-3-carboxamide (3f). Yield 55%, mp: 153–154 °C, MS-ESI (m/z): 371.1 [M+ + 1].; IR (KBr) (ν, cm−1): 3359 (NH), 3292 (OH), 1661 and 1631 (C
O); 1H NMR (400 MHz, DMSO-d6): δ 1.59–1.64 (m, 8H), 2.37 (s, 3H), 3.18–3.21 (t, J = 6.4 Hz, 2H), 3.43–3.47 (m, 6H), 4.41–4.43 (t, J = 5.6 Hz, 1H, OH), 6.92 (s, 1H), 7.31–7.33 (d, J = 8 Hz, 2H), 7.81–7.83 (d, J = 8.4 Hz, 2H), 8.12–8.15 (t, J = 5.6 Hz, 1H, NH).
4-(Dimethylamino)-N-(3-hydroxypropyl)-2-oxo-6-phenyl-2H-pyran-3-carboxamide (3g). Yield 45%, mp: 132–133 °C, MS-ESI (m/z): 316.9 [M+ + 1]; IR (KBr) (ν, cm−1): 3359 (NH), 3286 (OH), 1654 and 1630 (C
O); 1H NMR (400 MHz, DMSO-d6): δ 1.61–1.64 (t J = 6.4 Hz, 2H), 3.10 (s, 6H), 3.19–3.24 (q, J = 6 Hz, 2H), 3.43–3.48 (q, J = 5.6 Hz, 2H), 4.42–4.45 (t, J = 5.2 Hz, 1H, OH), 6.91 (s, 1H), 7.47–7.53 (m, 3H), 7.90–7.93 (m, 2H), 8.09–8.12 (t, J = 5.6 Hz, 1H, NH).
4-(Dimethylamino)-6-(4-fluorophenyl)-N-(3-hydroxypropyl)-2-oxo-2H-pyran-3-carboxamide (3h). Yield 42%, mp: 112–113 °C, MS-ESI (m/z): 335.2 [M+ + 1]; IR (KBr) (ν, cm−1): 3361 (NH), 3309 (OH), 1649 and 1624 (C
O); 1H NMR (400 MHz, DMSO-d6): δ 1.57–1.60 (t, J = 6.4 Hz, 2H), 3.12 (s, 6H), 3.18–3.23 (q, J = 6.8 Hz, 2H), 3.46–3.49 (t, J = 6.4 Hz, 2H), 4.46–4.49 (t, J = 5.2 Hz, 1H, OH), 6.89 (s, 1H), 7.31–7.34 (t, J = 8.6 Hz, 2H), 8.01–8.13 (m, 2H), 8.15–8.18 (t, J = 5.6 Hz, 1H, NH).
4-(Dimethylamino)-N-(3-hydroxypropyl)-2-oxo-6-(p-tolyl)-2H-pyran-3-carboxamide (3i). Yield 45%, mp: 86–87 °C, MS-ESI (m/z): 331.1 [M+ + 1]; IR (KBr) (ν, cm−1): 3355 (NH), 3291 (OH), 1651 and 1632 (C
O); 1H NMR (400 MHz, DMSO-d6): δ 1.57–1.64 (m, 2H), 2.49 (s, 3H), 3.09 (s, 6H), 3.19–3.22 (q, J = 6.4 Hz, 2H), 3.43–3.48 (q, J = 6.4 Hz, 2H), 4.31–4.44 (t, J = 5.2 Hz, 1H, OH), 6.85 (s, 1H), 7.32–7.34 (d, J = 8.3 Hz, 2H), 7.80–7.82 (d, J = 8 Hz, 2H), 8.07–8.10 (t, J = 5.2 Hz, 1H, NH).
3.2 Biological evaluations
For viral penetration assay prechilled (at 4 °C for 1 h) Vero cell monolayers were subsequently incubated with HSV-1F (300 pfu per well) for 3 h at 4 °C to allow viral adsorption. The infected cells were then incubated with a test compound (18.0 μg ml−1), DMSO (0.1%) or ACV (5.0 μg ml−1) for another 20 min at 37 °C to facilitate viral penetration. At the end of the incubation period, extracellular non-penetrated virus was inactivated by citrate buffer (pH 3.0) for 1 min, and then the cells were washed with PBS and overlaid with overlay medium for plaque formation. The viral plaques developed after 48 h of incubation at 37 °C were stained and counted.32
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g with a microcentrifuge for 10 min at 4 °C, subjected to SDS-PAGE and blotted with pre-equilibrated PVDF membrane (Thermo scientific, USA). Then the membrane was blocked in 5% NFDM in 1× TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Tween 20), rinsed and incubated with anti-HSV DNA polymerase or polyclonal anti-β-actin (Shanta Cruz Biotech Inc., USA) antibody in 5% BSA at 4 °C overnight. Immunoblotting was performed with peroxidase-labeled anti-rabbit polyclonal antibodies and visualized by an ECL Western blotting detection kit (Millipore, USA).34,35
000g with a microcentrifuge for 10 min at 4 °C, subjected to SDS-PAGE and blotted with pre-equilibrated PVDF membrane (Thermo scientific, USA). Then the membrane was blocked in 5% NFDM in 1× TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Tween 20), rinsed and incubated with anti-HSV DNA polymerase or polyclonal anti-β-actin (Shanta Cruz Biotech Inc., USA) antibody in 5% BSA at 4 °C overnight. Immunoblotting was performed with peroxidase-labeled anti-rabbit polyclonal antibodies and visualized by an ECL Western blotting detection kit (Millipore, USA).34,353.3 Molecular modeling
4. Summary and conclusions
The present study reveals the structure based optimization of preliminary lead compound II to a potential lead candidate 3d as a possible anti-HSV agent. A modeling study suggests that 3d can efficiently utilize tubular cavity-A for binding with HSV DNA polymerase and opens a future scope for drug candidate development. The newly synthesized compound 3d demonstrates the promising anti-HSV activity and biological studies were conducted to understand the possible mode of action. The MTT and plaque reduction assay revealed that 3d has moderate antiviral activity against HSV-1F and HSV-2G with respect to its EC50 and selectivity index, compared to ACV. The plaque reduction assay demonstrated that 3d inhibited HSV-1F and HSV-2G infection in a dose-dependent manner and >99% inhibition was achieved at 18.0 and 24.0 μg ml−1 respectively. In order to understand the quantitative and temporal aspects of the antiviral activity of 3d, we conducted kinetic studies, which revealed that the addition of 3d to virus infected cells at 4–6 h post-infection is highly effective in killing the virus. Further study showed that 3d was unable to prevent the attachment or penetration of HSV, like ACV, suggesting that the mode of action of 3d is not the prevention of viral adsorption or penetration, but perhaps the interference of early events of HSV replication. Moreover, the drug combination assay (3d ± ACV) showed strong synergistic effects, suggesting that 3d may work through similar targets but at different time points. Next, to investigate the possible mode of action of 3d on early events of the viral infection cycle, we studied the effect of 3d on IE gene expression of HSV. To study whether 3d interferes with any of the events of IE gene expression first we measured the two major end products, ICP4 and ICP27, of the IE gene by quantitative real-time PCR analysis. The results demonstrated the reduced expression of both ICP4 and ICP27 in HSV infected 3d treated cells but not in untreated cells. Furthermore, the depletion of viral DNA polymerase expression, demonstrated by immunoblotting studies, in HSV-1F infected cells treated with 3d indicated that it can prevent HSV multiplication.Acknowledgements
This research received funding from Department of Biotechnology (DBT), Delhi, India through grant no BT/PR14237/MED/29/196/2010 and BT/PR13759/PBD/17/686/2010. SK thanks CSIR, India for Senior Research Fellowships (SRF). The authors acknowledge Cosmic Discoveries Ltd. ILS, Hyderabad for NMR-Mass Facility, and Central Instrument Facility, BIT Mesra for analytical support.References
- A. J. Phillips, J. Biomed. Inf., 2006, 39, 18–33 CrossRef CAS.
- R. J. Brideau, M. L. Knechtel, A. Huang, V. A. Vaillancourt, E. E. Vera, N. L. Oien, T. A. Hopkins, J. L. Wieber, K. F. Wilkinson and B. D. Rush, Antiviral Res., 2002, 54, 19–28 CrossRef CAS.
- N. L. Oien, R. J. Brideau, T. A. Hopkins, J. L. Wieber, M. L. Knechtel, J. A. Shelly, R. A. Anstadt, P. A. Wells, R. A. Poorman and A. Huang, Antimicrob. Agents Chemother., 2002, 46, 724–730 CrossRef CAS.
- J. Piret and G. Boivin, Antimicrob. Agents Chemother., 2010, 55(2), 459–472 CrossRef.
- M. N. Prichard, E. R. Kern, C. B. Hartline, E. R. Lanier and D. C. Quenelle, Antimicrob. Agents Chemother., 2011, 55(10), 4728–4734 CrossRef CAS.
- J. L. Coleman and D. Shukla, Hum. Vaccines Immunother., 2013, 9(4), 729–735 CrossRef CAS.
- C. Johnston, D. M. Koelle and A. Wald, J. Clin. Invest., 2011, 121(12), 4600–4609 CAS.
- K. Modjarrad and S. H. Vermund, Lancet Infect. Dis., 2010, 10(7), 455–463 CrossRef.
- S. Karampuri, P. Bag, S. Yasmin, D. K. Chouhan, C. Bal, D. Mitra, D. Chattopadhyay and A. Sharon, Bioorg. Med. Chem. Lett., 2012, 22, 6261–6266 CrossRef CAS.
- C20H24N2O4, M = 356.4, triclinic, space group: P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.0084 (5), b = 11.0499 (7), c = 11.0645 (7) Å, a = 70.326 (5), b = 80.844 (5), g = 71.336 (6), V = 872.03 (10) Å3, T = 99.9 (2) K, Z = 2, m = 0.31 mm−1, F(000) = 377.4, Dc = 1.353 Mg m−1, yellow rectangular crystal, crystal size: 0.20 × 0.26 × 0.11 mm, 4704 reflections measured, 3038 unique, R1 =0.0438 for 2605Fo > 4sig(Fo) and 0.0509 for all 3038 data and 312 parameters with goodness of fit GooF = 1.079. Unit cell determination and intensity data collection (2θ range = 8–133.2°) was performed with 99.8% completeness at 99.9 (2) K. Structure solutions by direct methods and refinements by full-matrix least-squares methods on F2.
, a = 8.0084 (5), b = 11.0499 (7), c = 11.0645 (7) Å, a = 70.326 (5), b = 80.844 (5), g = 71.336 (6), V = 872.03 (10) Å3, T = 99.9 (2) K, Z = 2, m = 0.31 mm−1, F(000) = 377.4, Dc = 1.353 Mg m−1, yellow rectangular crystal, crystal size: 0.20 × 0.26 × 0.11 mm, 4704 reflections measured, 3038 unique, R1 =0.0438 for 2605Fo > 4sig(Fo) and 0.0509 for all 3038 data and 312 parameters with goodness of fit GooF = 1.079. Unit cell determination and intensity data collection (2θ range = 8–133.2°) was performed with 99.8% completeness at 99.9 (2) K. Structure solutions by direct methods and refinements by full-matrix least-squares methods on F2. - J. Meletiadis, S. Pournaras, E. Roilides and T. J. Walsh, Antimicrob. Agents Chemother., 2009, 54(2), 602–609 CrossRef.
- V. Petraitis, R. Petraitiene, W. W. Hope, J. Meletiadis, D. Mickiene, J. E. Hughes, M. P. Cotton, T. Stergiopoulou, M. Kasai, A. Francesconi, R. L. Schaufele, T. Sein, N. A. Avila, J. Bacher and T. J. Walsh, Antimicrob. Agents Chemother., 2009, 53, 2382–2391 CrossRef CAS.
- M. C. Berenbaum, Pharmacol. Rev., 1989, 41, 93–141 CAS.
- J. A. Hobden, M. Kumar, H. E. Kaufman, C. Clement, E. D. Varnell, P. S. Bhattacharjee and J. M. Hill, Invest. Ophthalmol. Visual Sci., 2010, 52(2), 830–833 Search PubMed.
- M. C. Berenbaum, J. Antimicrob. Chemother., 1987, 19, 271–273 CrossRef CAS.
- R. W. Honess and B. Roizman, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1276–1280 CrossRef CAS.
- J. P. Weir, Virus Genes, 1998, 16, 85–93 CrossRef CAS.
- C. A. Smith, P. Bates, R. Rivera-Gonzalez, B. Gu and N. A. DeLuca, J. Virol., 1993, 67, 4676–4687 CAS.
- C. A. Spencer, M. E. Dahmus and S. A. Rice, J. Virol., 1997, 71, 2031–2040 CAS.
- M. A. Hardwicke and R. M. Sandri-Goldin, J. Virol., 1994, 68, 4797–4810 CAS.
- J. S. Gibbs, H. C. Chiou, J. D. Hall, D. W. Mount, M. J. Retondo, S. K. Weller and D. M. Coen, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 7969–7973 CrossRef CAS.
- I. R. Lehman and P. E. Boehmer, J. Biol. Chem., 1999, 274, 28059–28062 CrossRef CAS.
- P. J. Vaughan, D. J. Purifoy and K. L. Powell, J. Virol., 1985, 53, 501–508 CAS.
- M. L. Gallo, D. I. Dorsky, C. S. Crumpacker and D. S. Parris, J. Virol., 1989, 63, 5023–5029 CAS.
- M. L. Gallo, D. H. Jackwood, M. Murphy, H. S. Marsden and D. S. Parris, J. Virol., 1988, 62, 2874–2883 CAS.
- Y. Zhang, P. P. But, V. E. Ooi, H. X. Xu, G. D. Delaney, S. H. Lee and S. F. Lee, Antiviral Res., 2007, 75, 242–249 CrossRef CAS.
- D. Ojha, H. Mukherjee, S. Ghosh, P. Bag, S. Mondal, N. Chandra, K. Mondal, S. Chakrabarti, A. Samanta and D. Chattopadhyay, J. Appl. Microbiol., 2013, 115, 1317–1328 CrossRef CAS.
- P. Bag, D. Chattopadhyay, H. Mukherjee, D. Ojha, N. Mandal, M. C. Sarkar, T. Chatterjee, G. Das and S. Chakraborti, Virol. J., 2012, 9, 98 CrossRef.
- D. Chattopadhyay, M. C. Sarkar, T. Chatterjee, R. Sharma Dey, P. Bag, S. Chakraborti and M. T. Khan, New Biotechnol., 2009, 25, 347–368 CrossRef CAS.
- H. Y. Cheng, T. C. Lin, C. M. Yang, K. C. Wang, L. T. Lin and C. C. Lin, J. Antimicrob. Chemother., 2004, 53, 577–583 CrossRef CAS.
- H. Mukherjee, D. Ojha, P. Bag, H. S. Chandel, S. Bhattacharyya, T. K. Chatterjee, P. K. Mukherjee, S. Chakraborti and D. Chattopadhyay, Microbiol. Res., 2013, 168, 238–244 CrossRef CAS.
- L. T. Lin, T. Y. Chen, C. Y. Chung, R. S. Noyce, T. B. Grindley, C. McCormick, T. C. Lin, G. H. Wang, C. C. Lin and C. D. Richardson, J. Virol., 2011, 85, 4386–4398 CrossRef CAS.
- J. Piret, S. Roy, M. Gagnon, S. Landry, A. Desormeaux, R. F. Omar and M. G. Bergeron, Antimicrob. Agents Chemother., 2002, 46, 2933–2942 CrossRef CAS.
- S. La Frazia, C. Amici and M. G. Santoro, Antiviral Ther., 2006, 11, 995–1004 CAS.
- P. Bag, D. Ojha, H. Mukherjee, U. C. Halder, S. Mondal, S. Nandi, A. Sharon, S. Chakrabarti and D. Chattopadhyay, PLoS One, 2013, 8(10), e77937 CAS.
- Schrödinger, Suite, LLC, New York, NY, 2013 Search PubMed.
- Schrödinger, LigPrep-V-2.7, LLC, New York, NY, 2013 Search PubMed.
- Schrödinger, Glide-V-6.0, LLC, New York, NY, 2013 Search PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS.
- Schrödinger, MacroModel-V-10.1, LLC, New York, NY, 2013 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 970219. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra01303d |
| ‡ These author contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |