
Modulated T carbon-like carbon allotropes: an ab initio study
Abstract
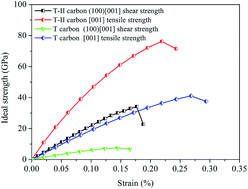
The structural stability, mechanical properties, and dynamical properties of T carbon-like structures were extensively studied by first-principles calculations using density functional theory. A novel modulated T carbon-like carbon allotrope (T-II carbon) is predicted by means of first principles calculations. This structure has 8 atoms in the unit cell, possesses the Pn![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, and can be derived by stacking up two T carbons together. T-II carbon is a semiconductor with band gap 0.88 eV and has a higher hardness (27 GPa) than that of T carbon (5.6 GPa). The calculations of ideal strength and the electron localization function indicate that T-II carbon has better ability to resist shear strain than T carbon.
m space group, and can be derived by stacking up two T carbons together. T-II carbon is a semiconductor with band gap 0.88 eV and has a higher hardness (27 GPa) than that of T carbon (5.6 GPa). The calculations of ideal strength and the electron localization function indicate that T-II carbon has better ability to resist shear strain than T carbon.
 Please wait while we load your content...
Please wait while we load your content...

