 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas phase hydration of amino acids and dipeptides: effects on the relative stability of zwitterion vs. canonical conformers
Ju-Young Kim
,
Doo-Sik Ahn†
,
Sung-Woo Park‡
and
Sungyul Lee
*
Department of Applied Chemistry, College of Applied Sciences, Kyung Hee University, Kyungki 446-701, S. Korea. E-mail: sylee@khu.ac.kr; Fax: +82-31-201-2340; Tel: +82-31-201-2423
First published on 14th March 2014
Abstract
We present a brief review of studies of the relative stability of canonical vs. zwitterionic forms of amino acids and dipeptides under the influence of gas phase hydration. Focus is given on the number of water molecules necessary to stabilize the zwitterionic conformer. Experimental and theoretical investigations for this interesting question are discussed. It is shown that the hydrating properties of amino acids and dipeptides are strongly dependent on the characteristics (hydrophilicity, basicity etc.) of side chains, the presence of metal cations, or an excess electron. Besides the relative Gibbs free energies of various conformers to estimate their relative thermodynamic stability, the activation barriers of proton transfer processes between canonical and zwitterionic forms are emphasized to assess the kinetic stability of thermodynamically less favorable species in low-temperature, gas phase environments.
 Ju-Young Kim | Ju-Young Kim was born in 1981 in Seoul, S. Korea. He received his BS (2008) and MS (2010) degree from Kyung Hee University, and is a PhD student in Department of Chemistry, the Graduate School, Kyung Hee University, under the guidance of Prof. Sungyul Lee. His research focus on mechanistic study of organocatalytic reactions, and theoretical study of gas phase hydration of biomolecules. |
 Doo-Sik Ahn | Doo-Sik Ahn received his BS (2003), MS (2005) degree from Kyung Hee university under the supervision of Prof. Sungyul Lee and PhD (2009) degree from Korea Advanced Institute of Science and Technology under the supervision of Prof. Sang Kyu Kim. He is now working as a research fellow at Fritz-Haber-Institute of the Max Planck Society. His research interests include the solvent effect on the bio-organic reactions, vibration mediated photodissociation dynamics, and the spectroscopic investigations of bimolecular ions embedded in liquid He droplets. |
 Sung-Woo Park | Sung-Woo Park received his BS (2000), MS (2004) and PhD (2010) degree from Kyung Hee university under the supervision of Prof. Sungyul Lee. During 2010–2013 he worked as a post doc at the Center for Superfunctional Materials in POSTECH. He is currently a research fellow in the Center for Catalytic Hydrocarbon Functionalization, Institute of Basic Science, Korea Institute of Science and Technology. His research interests include designing catalysts for activation of inert molecules and mechanistic studies of catalysis. |
 Sungyul Lee | Sungyul Lee earned his BS degree from Seoul National University in 1977 and his MS degree from KAIST in 1979. In 1988, he received his PhD degree in physical chemistry from the Department of Chemistry, University of Chicago under the supervision of Prof. K. F. Freed. He was appointed to Assistant, Associate, and Professor at Kyung Hee University from 1989 to present. His research interest is focused on the effects of water on the structures and reactivity of biomolecules, solvent catalysis (protic solvents, ionic liquids) of organic reactions, and materials chemistry. |
Introduction
Solvation is one of the most fundamental and interesting phenomena in chemical science. Although gas phase reactions proved to be instrumental for understanding chemical reactions in terms of molecular properties, the effects of solvents must ultimately be elucidated because most organic and biochemical reactions occur in solution phase. The role the solvent plays in chemical reactions is not limited to dissolving the reacting species. Interactions with solvent molecules influence the physicochemical properties of solutes, even tremendously increasing the rates of chemical reactions.Biochemical processes in cells, of course, provide many examples in which the solvent (water) profoundly affects the molecular structure and reactivity. Discussion of biochemical reactions is incomplete when omitting the role of water because water exerts such strong influence on the thermodynamics and kinetics of the processes. One prominent example is the structures of amino acids1–14 and peptides under the influence of water molecules. This interesting subject has been under intensive study, both theoretically and experimentally, because the structures and the stability of canonical15–18 and zwitterionic16,19–25 forms are profoundly affected by solvent. Amino acids exist in canonical forms in the gas phase, whereas, zwitterionic (charge-separated) conformer is predominant in aqueous solution.24–27 One of the central questions concerning the structures and biochemical properties of amino acids is: how many water molecules are required to stabilize the zwitterionic form? This question has been addressed by examining the relative stability of these two forms as a function of the number of interacting water molecules. Thus far, the experimental structural information on multiply hydrated amino acids is limited, and few previous studies have provided conclusive evidence as to how the structure of an amino acid changes upon solvation and where the transition from canonical structure to zwitterion occurs.
In this brief review, we discuss recent experimental and theoretical investigations for the role of water in determining the effects of gas phase hydration on relative stability of canonical (non-ionic) vs. zwitterionic forms of amino acids and dipeptides. We illustrate that both the thermodynamic and kinetic aspects of the chemistry of these biomolecules should be taken into account. The relevant parameters to be considered are the relative Gibbs free energies of various conformers and the activation barriers for transformations between them. Emphasis is placed on the latter property (Gibbs free energies of activation) to estimate the kinetic stabilities of biomolecules.
We illustrate that the role of water is profound: the interacting water molecules may either influence the structures of the conformers or take part in the transformation thereof directly (mediating the movement of the proton). We discuss the thermodynamic and kinetic stability of zwitterionic amino acids and dipeptides relative to canonical forms as a function of the number of water molecules, thereby, elucidating how many molecules are necessary to stabilize the zwitterion. It was identified that amino acids with different side chains (hydrophobic, acidic, basic, etc.) exhibit very different behavior with respect to how they are influenced by water. We also describe the effects of water on the canonical ↔ zwitterion transformation, especially its participation in multiple proton transfer processes. Our present review of amino acids and dipeptides is not exhaustive, limited to those for which attempts were made to unravel the influence of solvent on the stability of zwitterionic conformers. We present the case by classifying them based on the properties of their side chains. Amino acids and dipeptides with extra charges (with an extra electron, or metal cations) are also discussed. Studies of protonated amino acids would need separate discussion and are excluded from this brief review.
Glycine, alanine, valine: hydrophobic side chains
Effects of gas phase hydration on the structure of glycine28,29 (Gly) has been investigated by a number of experimentalists. Nonose et al.30 used electrospray ionization to study the structures and incremental binding energies of Gly. Kresin et al.31 investigated the hydroxyl loss fractions for Gly and tryptophan (Trp) molecules picked up by water clusters (H2O)n or (D2O)n (n ≤ 15) in a supersonic expansion and analyzed by electron bombardment mass spectrometry. Balabin32 reported the jet-cooled spontaneous Raman spectrum of a glycine–water complex (Gly(H2O)). The low-frequency vibrational spectrum (below 500 cm−1) of the solvated molecule is recorded and assigned using ab initio (MP2) and DFT (B3LYP, BLYP, PBE0 = PBE1PBE) methods. There are few experimental studies of the onset of the stabilized Gly zwitterion by explicit water molecules. Alonso et al.33 determined the structures of Gly(H2O) with a canonical Gly core by molecular-beam Fourier transform microwave spectroscopy. Bowen and co-workers8 studied hydrated Gly anions by mass spectrometric and size-selected photoelectron spectroscopy, finding that at least five water molecules are needed to transform a Gly anion into its zwitterion. By considering that an extra electron attached to an amino acid usually favors the formation of zwitterionic amino acids with fewer water molecules than do neutral amino acids, the authors concluded that at least five water molecules would be required to transform neutral Gly into its zwitterion. To date, the number of water molecules at the threshold of canonical → neutral transformation of Gly has not been determined experimentally.Aikens and Gordon's computational work9 on Gly(H2O)n (n = 0–8) is, to date, the most comprehensive theoretical study of gas phase hydration of Gly–water clusters. Employing a variety of quantum chemical methods, they suggested that Gly(H2O)7 with a zwitterionic Gly core is the global minimum energy structure, lying ∼1.3 kJ mol−1 below the lowest energy canonical conformer.
Thus, whereas the theoretical studies indicate that seven water molecules9 are required to stabilize the zwitterion on thermodynamic grounds, experimental observations8 for Gly–water clusters are not conclusive yet, only suggesting that at least five water molecules are necessary. Although the Gly(H2O)5–6 clusters with zwitterionic Gly cores are less stable (that is, have higher Gibbs free energies) than those with canonical Gly cores, the activation barriers to zwitterionic → canonical Gly(H2O)5–6 transformations may be of considerable interest. This is because the thermodynamically less favorable zwitterion may be kinetically stable (that is, they may exist for a long enough period of time to be observed experimentally in a low temperature (∼5 K) gas phase environment), if the barrier is sufficiently high. This may be the basis of Blom et al.'s7 experimental observation of zwitterionic Trp(H2O)5 whose Gibbs free energy was calculated to be 12.1 kJ mol−1 higher than the canonical counterpart, as discussed below.
Because the Gibbs free energy of Gly(H2O)5 with a zwitterionic Gly core presented in Fig. 1 is 6.7 kJ mol−1 higher than the canonical Gly(H2O)5,14c the formation of the former conformer is clearly less favorable. The zwitterion → canonical isomerization exhibits, on the other hand, a substantial barrier (36.8 kJ mol−1).14c This fairly large barrier may kinetically separate the zwitterionic and the canonical Gly(H2O)5, rendering them observable, at least in the gas phase, at low temperature.
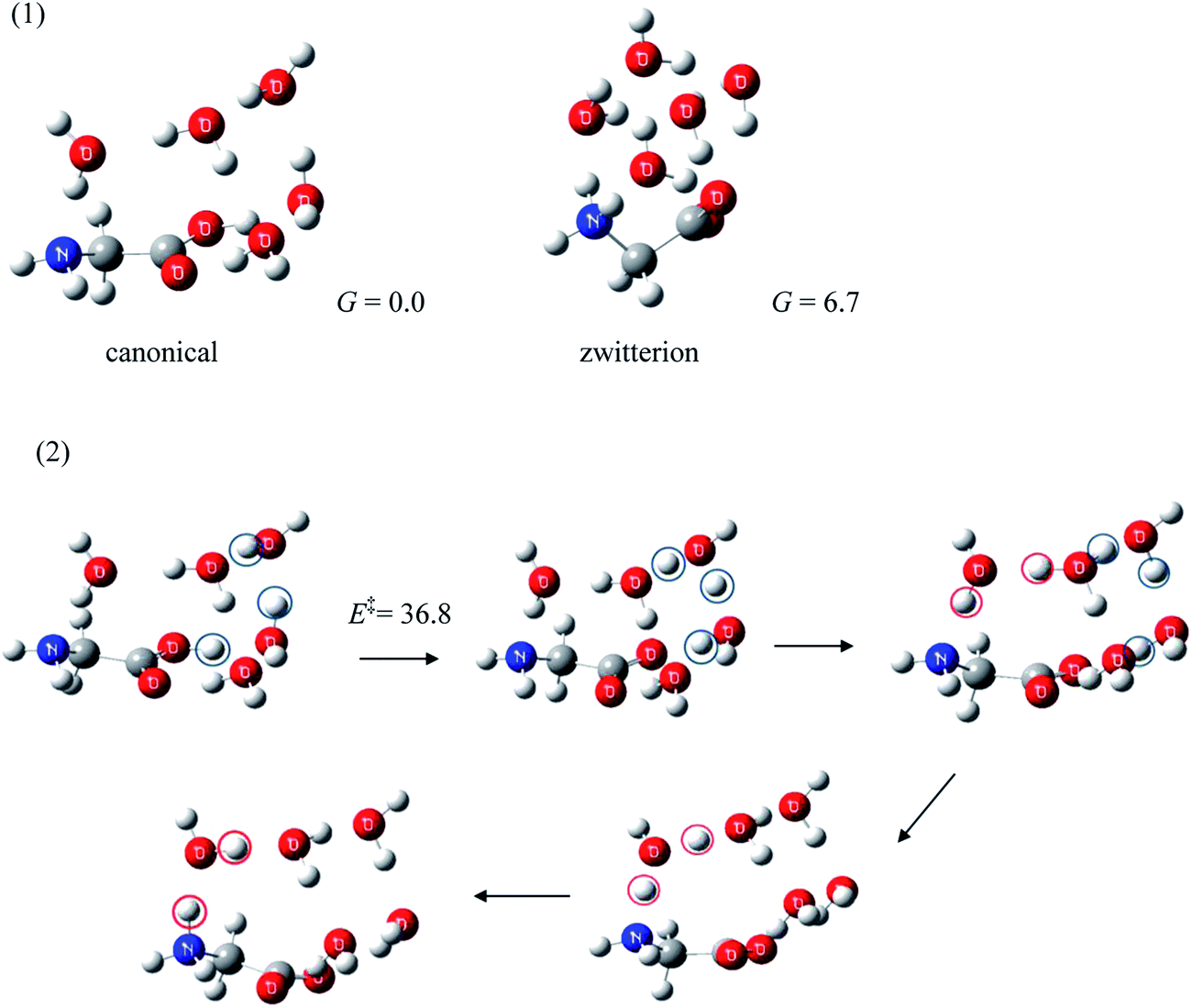 | ||
| Fig. 1 (1) Structures of the lowest energy conformers of canonical Gly(H2O)5 (relative Gibbs free energies in kJ mol−1, ZPE included; MP2/aug-cc-pvdz). (2) Dynamic pathway of zwitterion → canonical isomerization (reaction barrier in kJ mol−1, ZPE included; B3LYP/6-311++G(d,p)). | ||
There have been few investigations of this aspect of gas phase hydration of amino acids (zwitterionic ↔ canonical proton transfer processes of amino acid–water clusters) in comparison to the corresponding thermodynamic studies. To the best of our knowledge, the theoretical investigation of isomerization of Gly under the influence of up to eleven water molecules by Yamabe et al.34 seems to be the only study other than our work. They found that water molecules may act as a catalyst in proton transfer of Gly(H2O)4.
Many investigations were also carried out for Gly–water anions with an excess electron. Diken et al.35 found that Gly− and Gly−(H2O)1–2 have canonical Gly cores by photoelectron spectroscopy. Xu et al.8 observed that a zwitterionic species is formed for [Gly(H2O)n]− with n ≥ 5 based on mass spectrometry and photoelectron spectroscopy. Complexation with metal cations is also found to favor the formation of zwitterionic amino acids, decreasing the number of water molecules required. Williams and co-workers36 assessed the interactions between divalent metal ions and Gly. They found that, except for beryllium, metal ions render the Gly zwitterions more stable by 21–50 kJ mol−1 than Gly in its canonical form.
The situation for alanine (Ala) is similar to Gly in that the experimental observations of the Ala–water clusters are not conclusive, but theoretical studies clearly suggest that seven water molecules are required to stabilize the cluster with a zwitterionic Ala core. By examining the Ala(H2O)1–8 clusters employing a systematic approach (the EFP discrete solvation model with a Monte Carlo algorithm to sample the configuration space) to find the global minimum, Gordon and co-workers37 found that canonical and zwitterionic Ala(H2O)7 (Fig. 2) are quasidegenerate, either one being calculated to be the global minimum energy structure, depending on the theoretical methods.
 | ||
| Fig. 2 Structures of the lowest energy conformers of Ala(H2O)7 (relative energies in kJ mol−1). From ref. 37. | ||
The proton transfer process between zwitterionic and canonical Ala–water system has been studied only for Ala(H2O)2.15 Fig. 3 shows that isomerization proceeds by a concerted double proton transfer mediated by two water molecules. The barriers are, however, very small (<8.2 kJ mol−1), indicating the zwitterionic Ala(H2O)2, whose Gibbs free energies are higher than the canonical structures by >21 kJ mol−1, are, in addition, not kinetically stable.
 | ||
| Fig. 3 Concerted double proton transfer pathway from the Ala zwitterion–(H2O)2 clusters to the canonical clusters (reaction barriers in kJ mol−1). | ||
Calculations for the valine (Val)(H2O)n (n = 0–5) system by B3LYP/6-311++G(d,p), wB97XD/6-311++G(d,p) and MP2/aug-cc-pvdz level of theory revealed that five water molecules are necessary to stabilize the zwitterionic form of this amino acid.38 Fig. 4 shows the lowest energy canonical and zwitterionic forms of Val(H2O)5. Because the two forms are essentially quasidegenerate and the barrier of the canonical → zwitterionic pathway is appreciable, it seems that both forms of Val(H2O)5 may be observed at low temperature in the gas phase.38
 | ||
| Fig. 4 (1) Lowest-lying conformers of canonical Val(H2O)5 (relative Gibbs free energies in kcal mol−1; (a) wB97XD/6-311++G(d,p) (b) MP2/aug-cc-pvdz). (2) Transformation from the lowest energy zwitterionic (zw-5-1) and canonical (ca-5-1) conformer of Val(H2O)5 (relative energy in kcal mol−1, ZPE included; wB97XD/6-311++G(d,p)). | ||
The effects of hydration of Val–alkali metal ion complexes, Val·M+(H2O)n, (n = 2–6, M = Li, Na, and K), were probed by Williams and co-workers39 using both theory and blackbody infrared radiative dissociation experiments. They showed that Val·Li+(H2O)3 and Val·Na+(H2O)2 complexes are zwitterionic, indicating that the size of the metal cation may influence the gas phase hydration of Val.
Serine: hydrophilic side chain
Serine40 (Ser) is the smallest amino acid with a hydrophilic side chain. The hydroxyl group may participate in proton transfer in the neutral/zwitterion isomerization processes as an “intramolecular solvent”. The hydrophilic side chain, –OH, may also form hydrogen bonds with water molecules. Thus, assuming that proton transfer from the carboxyl to the amino group in Ser may involve as many water molecules as in the amino acids with hydrophobic side chains, it may be presumed that more water molecules would be necessary to stabilize the zwitterionic Ser. To date, gas phase hydration of Ser has been investigated only up to Ser(H2O)2.41 Since the energies of Ser(H2O)2 with zwitterionic Ser are at least 25 kJ mol−1 higher than the lowest energy canonical conformer (C3-2-a) shown in Fig. 5, they are considered to be unstable thermodynamically. Most of the zwitterionic Ser(H2O)2 molecules isomerize to canonical forms without significant barriers, making them also kinetically unstable. Some may, however, isomerize to the canonical form via considerable barriers, such as the zwitterionic (Z2-6) whose barrier is 26.0 kJ mol−1. | ||
| Fig. 5 (1) Lowest-lying conformers of canonical Ser(H2O)2 (relative energies in kJ mol−1, ZPE included; B3LYP/6-31++G(d,p), (a) MP2/6-31++G(d,p)). (2) Mechanism of canonical ↔ zwitterion isomerization of Ser(H2O)2 (reaction barrier in kJ mol−1, ZPE included; B3LYP/6-31++G(d,p)). | ||
Arginine, lysine, histidine: basic side chains
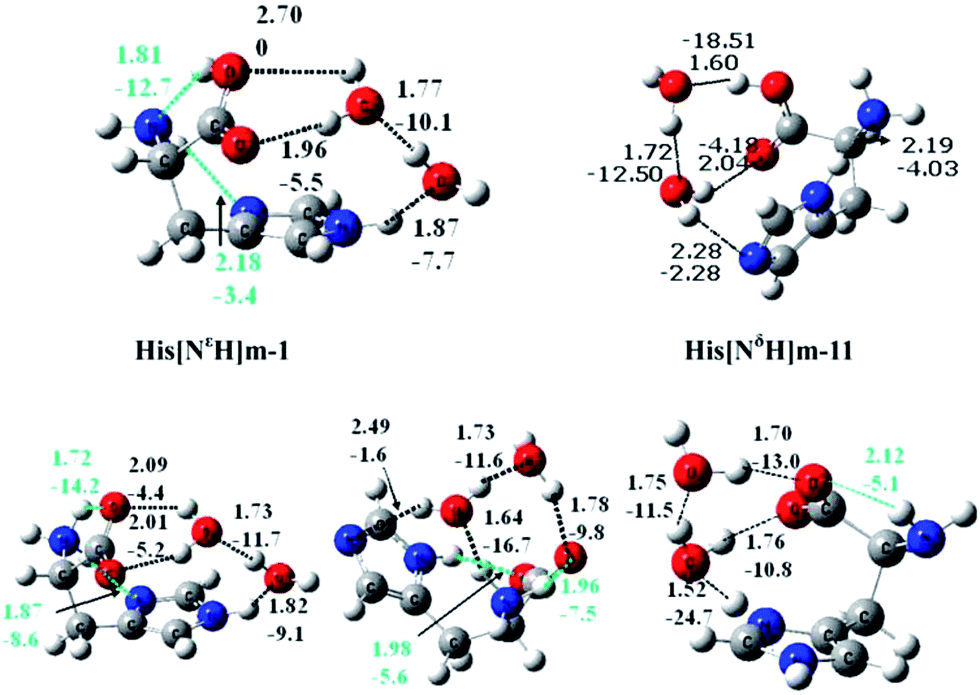
Saykally and co-workers42 were the first to examine the relative stability of zwitterionic/canonical arginine43–48 (Arg) in the gas phase. By employing the infrared cavity ringdown laser absorption spectroscopy, they clearly observed that the conformer of gas phase Arg is canonical. Although Arg is similar to other amino acids in that the canonical form is only observed in the gas phase, the presence of the strongly basic guanidine side chain in Arg may confer properties that are distinct from the other amino acids. For example, this type of side chain may render the proton transfer from the carboxyl group more facile under the influence of solvent. So far, however, no experimental study of Arg–water system concerning the relative stability of zwitterionic/canonical Arg has been reported.Fig. 6 presents the calculated structures and relative energy of the zwitterionic and canonical conformers of Arg(H2O).49 The energy of the conformer (Z22-1) with the zwitterionic Arg core is lower than that of the lowest energy canonical form (C5-1) by ∼7.0 kJ mol−1 by the B3LYP/6-311++G(d,p) method. This observation is quite striking, considering that the zwitterionic Arg is at least 8 kJ mol−1 higher in energy than the canonical Arg.49 It is also remarkable to observe that a single water molecule may stabilize the zwitterionic Arg relative to the canonical form because the consensus had been that at least five molecules of water are necessary to make the zwitterionic form energetically competitive with the canonical conformer. It must be noted that a proton is transferred from the carboxyl to the side chain guanidine group in Arg, in contrast with other amino acids. The two zwitterionic conformers (Z22-1) and (Z21-2), depicted in Fig. 6, are almost of the same energy (within 2.4 kJ mol−1).
 | ||
| Fig. 6 Structures of the lowest energy conformers of (1) zwitterionic and (2) canonical Arg(H2O) (relative energies with respect to (Z22-1) in kJ mol−1, ZPE included; (a) B3LYP/6-31++G(d,p) and (b) MP2/aug-cc-pvdz and bond lengths in Å). | ||
One important issue is the mechanism and the barrier of formation of the zwitterionic Arg(H2O) from the canonical conformer. If the Arg(H2O) complex initially produced with a canonical Arg core is kinetically very stable (that is, if the barrier to zwitterionic Arg(H2O) is high), the latter form of Arg(H2O) could hardly be formed. The barrier from the canonical form to the zwitterion is ∼18.7 kJ mol−1 (Fig. 7), indicating that the canonical conformer of Arg(H2O), once formed, may easily transform to the zwitterion. Fig. 7 also shows that the lowest energy zwitterionic form (Z22-1) converts via a double proton transfer process to the canonical form.
 | ||
| Fig. 7 Isomerization from the canonical form of Arg(H2O) to the zwitterionic form (relative energies, relative Gibbs free energies and reaction barrier in kJ mol−1, ZPE included; (a) B3LYP/6-311++G(d,p) and (b) MP2/6-311+G(d)). | ||
Effects of metal cations on the relative stability of zwitterionic/canonical Arg were studied extensively by Williams and co-workers.45,50,51 They observed that cationized Arg45,52 (Arg·M+, M = H. Li, Na, K, Rb, Cs) is in the zwitterionic form and that one water molecule stabilizes the cationized Arg.50 An excess electron tends to stabilize the zwitterionic conformer relative to the canonical forms. For example, Gutowski and co-workers52 reported that Arg solvated by an excess electron renders the zwitterionic and canonical Arg quasidegenerate. The effects of anions on the structure of Arg have been treated very recently by Milner et al.53 These authors found that fragmentation of X−·Arg (X− = F−, Cl−, Br−, I−, NO3−, ClO3−) produces the Arg zwitterion.
The side chain of lysine (Lys) is very basic, second only to Arg among the natural amino acids. Theoretical work, carried out in our lab,54 examined Lys(H2O)n (n = 2, 3), predicting that zwitterionic Lys becomes quasidegenerate with the canonical forms due to the solvating effects of three water molecules. This smaller number of water molecules to stabilize the Lys zwitterion than that (>5 water molecules) for the amino acids with hydrophobic side chains, described above, seems to be the effect of the strongly basic side chain of Lys. Fig. 8 depicts the structures of Lys(H2O)3 with zwitterionic and canonical Lys cores, in which the Gibbs free energy of the zwitterionic conformer is shown to be lower than that of the canonical form.
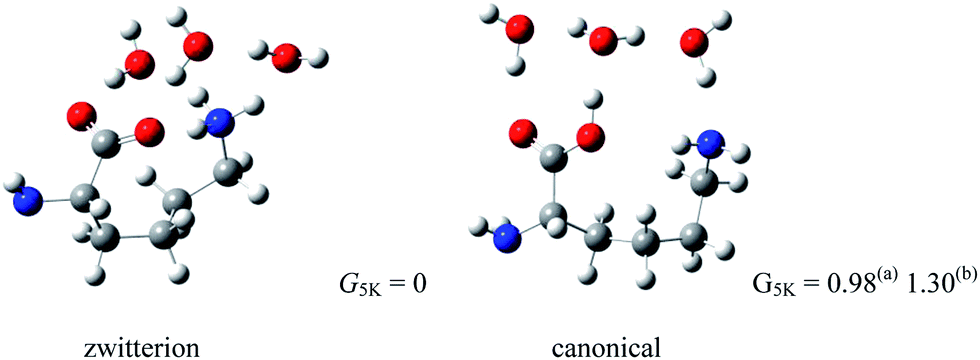 | ||
| Fig. 8 Structures of lowest energy canonical and zwitterionic forms of Lys(H2O)3 (relative Gibbs free energies in kJ mol−1, ZPE included; (a) MP2/aug-cc-pvdz and (b) wB97XD/6-311++G(d,p)). | ||
Effects of metal cations on the stability of zwitterionic Lys were studied by Williams group.55–57 By using density functional theory and blackbody infrared radiative dissociation experiments, they found that the lowest-energy structure of lithiated Lys without a water molecule is nonzwitterionic; adding a water molecule to lithiated Lys did not stabilize the zwitterionic form of Lys. The Lys·Na+ and Lys·K+ complexes determined by infrared multiple photon dissociation spectroscopy were also reported to be nonzwitterionic.56 The related amino acids α-N-methyllysine and ε-N,N-dimethyllysine, however, were shown to be zwitterionic57 when complexed with Na+ or K+, and with Li+, Na+, or K+, respectively, illustrating that different side chains can have very different effects on the stability of different conformers in the metal cationized amino acids.
Gas phase hydration of histidine (His) was treated by Lin and co-workers.58 Their calculations showed that for His(H2O)1–2 the canonical forms are clearly more stable than the zwitterion (Fig. 9). Effects of metal cations on the conformation of His was studied by Armentrout and co-workers.59
 | ||
| Fig. 9 Structures of lowest energy conformers of His(H2O)2 (bond lengths in Å). From ref. 59. | ||
Proline: cyclic amino acid
The structure of proline60,61 (Pro) is somewhat different from that of other amino acids. Due to the embedded pyrrolidine ring, Pro plays an important role in the formation of the β-turn structure in polypeptides and proteins.62 Pro is also unique among the amino acids in that the N-terminus is a secondary amine. Because it is well known that secondary amines exhibit both greater basicity (in aqueous solution) and proton affinity (in the gas phase) than do primary amines,63 this structural feature of Pro may show the effects of gas phase hydration that are different from that of other amino acids with primary amine groups. The observation of sodiated Pro zwitterion by Ohanessian and co-workers64 in the gas phase seems to be the result of this effect.Fig. 10 depicts the structures of Pro(H2O)4 optimized by wB97XD/6-311++G(d,p) and MP2/aug-cc-pvdz methods.65 Because the relative Gibbs free energy G5 K of Pro(H2O)4 with a canonical Pro core is higher than that with zwitterionic Pro by 1.3 (4.6) kJ mol−1 at wB97XD/6-311++G(d,p) (MP2/aug-cc-pvdz) level of theory, four water molecules seem to be enough to stabilize the Pro zwitterion.
 | ||
| Fig. 10 Structures of lowest energy canonical and zwitterionic forms of Pro(H2O)4 (relative Gibbs free energies in kJ mol−1, ZPE included; (a) wB97XD/6-311++G(d,p) and (b) MP2/aug-cc-pvdz). | ||
Tryptophan, phenylalanine: aromatic side chain
Because of the presence of the aromatic rings, gas phase hydration of Trp and phenylalanine (Phe) are more amenable to studies by ultraviolet (UV) spectroscopy than other amino acids lacking the appropriate chromophore. The early pioneering UV spectroscopic work on singly hydrated neutral tryptophan was carried out using resonance-enhanced two-photon ionization and laser-induced fluorescence spectroscopy. Peteanu and Levy66 investigated resonant two-photon ionization spectrum of singly hydrated Trp. Sulkes et al.67 used laser-induced fluorescence to examine the excitation spectrum of the Trp–single water complex. Simons and co-workers68 investigated the hydrated complexes of Trp by a combination of calculations, ion dip infrared spectroscopy, UV hole burning, and resonant two-photon ionization. Bowen and co-workers' experiments8 on the anions of Trp set four water molecules as lower limits for the number of water molecules needed to induce zwitterion formation in these amino acids. It was Paizs, Oomens and co-workers7 that unambiguously determined the onset of Trp zwitterion under the influence of water molecules. By means of the IR spectroscopy and DFT calculations for Trp(H2O)1–6 complexes, they observed that the zwitterionic Trp may be stabilized by four water molecules. These authors obtained the IR spectra and assigned the bands at 1300–1850 cm−1 to either canonical or zwitterionic Trp conformers. They also observed weak absorption by zwitterionic Trp(H2O)5 and Trp(H2O)6. It would be interesting to interpret these results in terms of thermodynamic and kinetic stability of the zwitterionic vs. canonical Trp as dictated by the relative Gibbs free energy and the activation barrier of zwitterion → canonical transformation process, respectively. Because the calculated energy of zwitterionic Trp(H2O)5 is higher by 12.1 kJ mol−1 than that of the canonical form, the weak absorption by zwitterionic Trp(H2O)5 may not be explained by the energy gap, as the authors pointed out. Thus, it may be that a modest barrier for zwitterion → canonical change renders a zwitterionic Trp(H2O)5 kinetically stable at low temperature in the gas phase. For Trp(H2O)6, the energy gap narrows down to ∼4 kJ mol−1 (canonical form still more stable), so that the minor absorption may readily be assigned as that of zwitterionic Trp(H2O)6.As for Phe, Bowen and co-workers8 suggested that the zwitterionic form of Phe may be stabilized by four water molecules.
Diglycine, dialanine, diproline: dipeptides
Considering that diglycine (Gly)2 is the simplest dipeptide, it is surprising to see that its gas phase hydration has not been studied. To the best of our knowledge, the (Gly)2–water system has not yet been examined experimentally. Several reports exist for protonated69,70 and cationized (Gly)2.71 Using infrared multiple photon dissociation (IRMPD) spectroscopy and MP2(full)/6-311++G(2d,2p)//B3LYP/6-311+G(d,p) level of theory, Wu and McMahon69 observed that the most stable (Gly)2H+ is canonical (Fig. 11). Fig. 12 compares the Gibbs free energies of (Gly)2(H2O)3 with canonical and zwitterionic (Gly)2 core recently obtained in a computational study in our lab.72 Because the Gibbs energy of zwitterionic (Gly)2(H2O)3 is 31.6 kJ mol−1 higher than that of canonical (Gly)2(H2O)3, it is predicted that hydration by up to three water molecules does not give thermodynamic stability of the zwitterion relative to the canonical forms. Our calculations also suggest that zwitterionic (Gly)2(H2O)3 is not stable kinetically. | ||
| Fig. 11 Structures and IRMPD spectra of lowest energy conformers of (1) (Gly)2H+ and (2) (Pro)2H+. From ref. 69. | ||
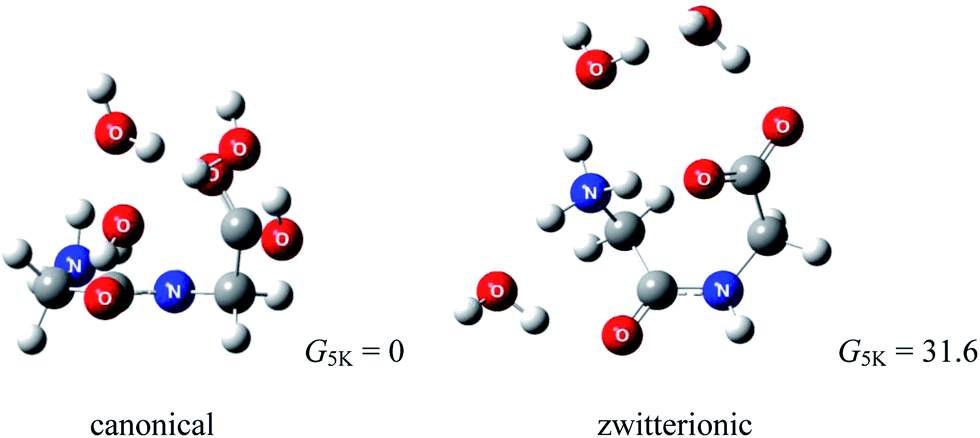 | ||
| Fig. 12 Structures of lowest energy canonical and zwitterionic forms of (Gly)2(H2O)3 (relative Gibbs free energies in kJ mol−1, ZPE included; wB97XD/6-311++G(d,p)). | ||
Although conformers of dialanine (Ala)2 in water have been studied by several groups using various theoretical methods,73–75 to the best of our knowledge, no study has been reported for its gas phase hydration. For the protonated diproline (Pro)2H+, Wu and McMahon69 reported that the lowest energy structure of (Pro)2H+ is zwitterionic, although both canonical and zwitterionic forms were detected in their infrared multiple photon dissociation experiments (Fig. 11).
Conclusions
In the present review, we discussed the important role water plays in affecting the structures of amino acids and dipeptides. We showed that the solvent water may not only serve as an electrostatic continuum, but it may also act as a direct participant in proton transfer, mediating, and/or promoting the process. Further experimental studies for this extremely interesting system will be highly desirable.Acknowledgements
We thank the National Research Foundation of Korea (NRF-2012R1A2A2A02013289, NRF-2011-0021836) for financial support and the KISTI Supercomputing Center (2013).References
- C. Desfrancois, S. Carles and J. P. Schermann, Chem. Rev., 2000, 100, 3943 CrossRef CAS
.
- R. M. Balabin, J. Phys. Chem. Lett., 2010, 1, 20 CrossRef CAS
- A. G. Csaszar and A. Perczel, Prog. Biophys. Mol. Biol., 1999, 71, 243 CrossRef CAS
- F. Rogalewicz, G. Ohanessian and N. J. Gresh, Comput. Chem., 2000, 21, 963 CrossRef CAS
- P. D. Godfrey, S. Firth, L. D. Hatherley, R. D. Brown and A. P. Pierlot, J. Am. Chem. Soc., 1993, 115, 9687 CrossRef CAS
- X. Gao and G. Fischer, J. Phys. Chem. A, 1999, 103, 4404 (Spectrochim. Acta, 1999, 55, 2329) Search PubMed
- M. N. Blom, I. Compagnon, N. C. Polfer, G. von Helden, G. Meijer, S. Suhai, B. Paizs and J. Oomens, J. Phys. Chem. A, 2007, 111, 7309 CrossRef CAS
- S. Xu, J. M. Niles and K. H. Bowen, J. Chem. Phys., 2003, 119, 10696 CrossRef CAS
- C. M. Aikens and M. S. Gordon, J. Am. Chem. Soc., 2006, 128, 12835 CrossRef CAS
- I. Mayer and P. J. Valiron, Chem. Phys., 1998, 109, 3360 CAS
- T. S. Zwier, J. Phys. Chem. A, 2001, 105, 8827 CrossRef CAS
- L. C. Snoek, E. G. Robertson, R. T. Kroemer and J. P. Simons, J. Phys. Chem. Solids, Lett. Sect., 2001, 321, 49 CrossRef
- I. Compagnon, F. C. Hagemeister, R. Antoine, D. Rayane, M. Broyer, P. Dugourd, R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 2001, 123, 8440 CrossRef CAS
-
(a) D.-S. Ahn, A.-R. Kang, S. Lee, B. Kim, S. K. Kim and D. Neuhauser, J. Chem. Phys., 2005, 122, 084310 CrossRef
-
(a) D.-S. Ahn, S.-W. Park, I.-S. Jeon, M. K. Lee, N.-H. Kim, Y.-H. Han and S. Lee, J. Phys. Chem. B, 2003, 107, 14109 CrossRef CAS
- I.-S. Jeon, D.-S. Ahn, S.-W. Park, S. Lee and S. K. Kim, Chem. Phys. Lett., 2005, 403, 72 CrossRef CAS
- H. H. Jensen and M. S. Gordon, J. Am. Chem. Soc., 1993, 117, 8159 CrossRef
- S. J. Xu, W. J. Zheng, D. Radisic and K. H. Bowen, J. Chem. Phys., 2005, 122, 091103 CrossRef PubMed
- L. C. Snoek, R. T. Kroemer, M. R. Hockridge and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1819 RSC
- J. Spinor and M. Sulkes, J. Chem. Phys., 1993, 98, 9389 CrossRef
- Y. Ding and K. Krogh-Jespersen, Chem. Phys. Lett., 1992, 199, 261 CrossRef CAS
- A. Fernandez-Ramos, Z. Smedarchina, W. Siebrand and M. Z. Zgierski, J. Chem. Phys., 2000, 113, 9714 CrossRef CAS
- A. S. Lemoff, M. F. Bush and E. R. Williams, J. Phys. Chem. A, 2005, 109, 1903 CrossRef CAS PubMed
- C. H. Hu, M. Shen and H. F. Schafer, III, J. Am. Chem. Soc., 1993, 115, 2923 CrossRef CAS
- P. Bandyopadhyay, M. S. Gordon, B. Mennucci and J. Tomasi, J. Chem. Phys., 2002, 116, 5023 CrossRef CAS
- R. R. Julian and M. F. Jarrold, J. Phys. Chem. A, 2004, 108, 10861 CrossRef CAS
- J. H. Jensen and M. S. Gordon, J. Am. Chem. Soc., 1995, 117, 8159 CrossRef CAS
- L. C. Snoek, R. T. Kroemer and J. P. Simons, Phys. Chem. Chem. Phys., 2002, 4, 2130 RSC
- E. Kassab, J. Langlet, E. Evleth and Y. Akacem, J. Mol. Struct.: THEOCHEM, 2000, 531, 267 CrossRef CAS
- S. Nonose, S. Iwaoka, K. Mori, Y. Shibata and K. Fuke, Eur. Phys. J. D, 2005, 34, 315 CrossRef CAS
- R. Moro, R. Rabinovitch and V. V. Kresin, J. Chem. Phys., 2005, 123, 074301 CrossRef PubMed
- R. M. Balabin, J. Phys. Chem. B, 2010, 114, 15075 CrossRef CAS PubMed
- J. L. Alonso, E. J. Cocinero, A. Lesarri, M. E. Sanz and J. C. Lopez, Angew. Chem., Int. Ed., 2006, 45, 3471 CrossRef CAS PubMed
- S. Yamabe, N. Ono and N. Tsuchida, J. Phys. Chem. A, 2003, 107, 7915 CrossRef CAS
- E. G. Diken, N. I. Hammer and M. A. Johnson, J. Chem. Phys., 2004, 120, 9899 CrossRef CAS PubMed
- E. F. Strittmatter, A. S. Lemoff and E. R. Williams, J. Phys. Chem. A, 2000, 104, 9793 CrossRef CAS PubMed
- J. M. Mullin and M. S. Gordon, J. Phys. Chem. B, 2009, 113, 8657 CrossRef CAS PubMed
- J.-Y. Kim, G.-Y. Won and S. Lee, Bull. Korean Chem. Soc., 2012, 33, 3797 CrossRef CAS
- R. A. Jockusch, A. S. Lemoff and E. R. Williams, J. Phys. Chem. A, 2001, 105, 10929 CrossRef CAS
- S. Blanco, M. E. Sanz, J. C. Lopez and J. L. Alonso, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20183 CrossRef CAS PubMed
- I.-S. Jeon, D.-S. Ahn, S.-W. Park, S. Lee and B. Kim, Int. J. Quantum Chem., 2005, 101, 55 CrossRef CAS
- C. J. Chapo, J. B. Paul, R. A. Provencal, K. Roth and R. J. Saykally, J. Am. Chem. Soc., 1998, 120, 12956 CrossRef CAS
- R. J. Gdanitz, W. Cardoen, T. L. Windus and J. Simons, J. Phys. Chem. A, 2004, 108, 515 CrossRef CAS
- R. R. Julian, R. Hodyss and J. L. Beauchamp, J. Am. Chem. Soc., 2001, 123, 3577 CrossRef CAS PubMed
- R. A. Jockusch, W. D. Price and E. R. Williams, J. Phys. Chem. A, 1999, 103, 9266 CrossRef CAS PubMed
- M. F. Bush, J. T. O'Brien, J. S. Prell, R. J. Saykally and E. R. Williams, J. Am. Chem. Soc., 2007, 129, 1612 CrossRef CAS PubMed
- J. Rak, P. Skurski, J. Simons and M. Gutowski, J. Am. Chem. Soc., 2001, 123, 11695 CrossRef CAS PubMed
- W. D. Price, R. A. Jockusch and E. R. Williams, J. Am. Chem. Soc., 1997, 119, 11988 CrossRef CAS PubMed
- S. Im, S.-W. Jang, S. Lee, Y. Lee and B. Kim, J. Phys. Chem. A, 2008, 112, 9767 CrossRef CAS PubMed
- M. F. Bush, J. S. Prell, R. J. Saykally and E. R. Williams, J. Am. Chem. Soc., 2007, 129, 13544 CrossRef CAS PubMed
- M. W. Forbes, M. F. Bush, N. C. Polfer, J. Oomens, R. C. Dunbar, E. R. Williams and R. A. Jockusch, J. Phys. Chem. A, 2007, 111, 11759 CrossRef CAS PubMed
- P. Skurski, J. Rak, J. Simons and M. Gutowski, J. Am. Chem. Soc., 2001, 123, 11073 CrossRef CAS PubMed
- E. M. Milner, M. G. D. Nix and C. E. H. Dessent, J. Phys. Chem. A, 2012, 116, 801 CrossRef CAS PubMed
- T.-K. Hwang, G.-Y. Eom, M.-S. Choi, S.-W. Jang, J.-Y. Kim, S. Lee, Y. Lee and B. Kim, J. Phys. Chem. B, 2011, 115, 10147 CrossRef CAS PubMed
- A. S. Lemoff, M. F. Bush, J. T. O'Brien and E. R. Williams, J. Phys. Chem. A, 2006, 110, 8433 CrossRef CAS PubMed
- M. F. Bush, M. W. Forbes, R. A. Jockusch, J. Oomens, N. C. Polfer, R. J. Saykally and E. R. Williams, J. Phys. Chem. A, 2007, 111, 7753 CrossRef CAS PubMed
- M. F. Bush, J. Oomens and E. R. Williams, J. Phys. Chem. A, 2009, 113, 431 CrossRef CAS PubMed
- A. K. Rai, W. Fei, Z. Wu and Z. Lin, Theor. Chem. Acc, 2009, 124, 37 CrossRef CAS
- M. Citir, C. S. Hinton, J. Oomens, J. D. Steill and P. B. Armentrout, J. Phys. Chem. A, 2012, 116, 1532 CrossRef CAS PubMed
- A. Lesarri, S. Mata, E. J. Cocinero, S. Blanco, J. C. Lopez and J. L. Alonso, Angew. Chem., Int. Ed., 2002, 41, 4673 CrossRef CAS PubMed
- S. Mata, V. Vaquero, C. Cabezas, I. Pena, C. Perez, J. C. Lopez and J. L. Alonso, Phys. Chem. Chem. Phys., 2009, 11, 4141 RSC
- F. A. Momany, R. F. McGuire, A. W. Burgess and H. A. Scheraga, J. Phys. Chem., 1975, 79, 2361 CrossRef CAS
- H. Umeyama and K. Morokuma, J. Am. Chem. Soc., 1976, 98, 4400 CrossRef CAS
- C. Kapota, J. Lemaire, P. Maitre and G. Ohanessian, J. Am. Chem. Soc., 2004, 126, 1836 CrossRef CAS PubMed
- J.-Y. Kim and S. Lee, unpublished.
- L. A. Peteanu and D. A. Levy, J. Phys. Chem., 1988, 92, 6554 CrossRef CAS
- C. K. Teh, J. Sipior and M. Sulkes, J. Phys. Chem., 1989, 93, 5393 CrossRef CAS
-
(a) P. Carcabal, R. T. Kroemer, L. C. Snoek, J. P. Simons, J. M. Bakker, I. Compagnon, G. Meijer and G. von Helden, Phys. Chem. Chem. Phys., 2004, 6, 4546 RSC
- R. Wu and T. B. McMahon, J. Am. Chem. Soc., 2007, 129, 4864 CrossRef CAS PubMed
- R. Wu and T. B. McMahon, J. Phys. Chem. B, 2009, 113, 8767 CrossRef CAS PubMed
- M. M. Kish, C. Wesdemiotis and G. Ohanessian, J. Phys. Chem. B, 2004, 108, 3086 CrossRef CAS
- J.-Y. Kim, G.-Y. Won and S. Lee, Bull. Korean Chem. Soc., 2014, 35, 798 CrossRef CAS
- A. N. Drozdov, A. Grossfield and R. V. Pappu, J. Am. Chem. Soc., 2004, 126, 257 CrossRef PubMed
- Y. Yonezawa, I. Fukuda, N. Kamiya, H. Shimoyama and H. Nakamura, J. Chem. Theory Comput., 2011, 7, 1484 CrossRef CAS
- V. Cruz, J. Ramos and J. Martínez-Salazar, J. Phys. Chem. B, 2011, 115, 4880 CrossRef CAS PubMed
Footnotes |
| † Present address: Department of Chemistry, KAIST, Daejeon 305-701, S. Korea. |
| ‡ Present address: Fritz-Haber-Institute of Max Planck Society, Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2014 |