
Dual-sulfonated dipyridinium phosphotungstate catalyst for liquid-phase Beckmann rearrangement of cyclohexanone oxime
Abstract
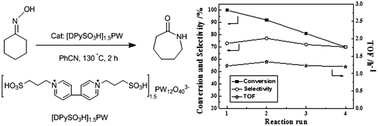
The heteropolyanion (HPA)-based ionic liquid (IL) hybrid was prepared by pairing Keggin-structured HPA of the phosphotungstate anion PW12O403− (PW) with dual-sulfonated 4,4′-dipyridinium IL-cation [DPySO3H]2+. The obtained powdered IL–HPA hybrid material [DPySO3H]1.5PW was characterized and catalytically assessed in Beckmann rearrangement of cyclohexanone-oxime to ε-caprolactam. Under the optimized reaction conditions: 10 mol% catalyst, 130 °C, 2 h, benzonitrile as the solvent, and without the environmentally harmful cocatalyst ZnCl2, [DPySO3H]1.5PW exhibits 100% conversion and 73.0% selectivity, remarkably higher than control catalysts involving other HPA-anions or the partially proton-substituted IL-cation. Moreover, the catalyst [DPySO3H]1.5PW can be isolated and reused via filtration from the liquid–solid heterogeneous reaction mixture demonstrated by the four-run recycling test.
 Please wait while we load your content...
Please wait while we load your content...

