 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanoparticles: calixarene complexation in a mixed calixarene–alkanethiol monolayer†
Petri M. S. Pulkkinena,
Jukka Hassinenb,
Robin H. A. Rasb and
Heikki Tenhu*a
aLaboratory of Polymer Chemistry, Department of Chemistry, University of Helsinki, P.O.Box 55, 00014 Helsinki, Finland. E-mail: heikki.tenhu@helsinki.fi
bDepartment of Applied Physics, Aalto University, Puumiehenkuja 2, FI-02150 ESPOO, Finland
First published on 28th February 2014
Abstract
This paper describes a detailed study on the complexation of pyridinium derivatives with calixarenes bound to gold nanoparticles (AuNPs). The studied calixarene derivatives are mixed with alkanethiols to form mixed monolayers on AuNP surfaces. The key findings are: (i) even a small amount (less than 11 mol%) of calixarenes can retain their complexation abilities among a majority of alkanethiols in a mixed monolayer, showing that it is possible to dilute the active calixarene (and possibly other receptors) in gold surfaces, (ii) the chain length of the alkanethiol compared with the calixarene spacer length can be used to fine tune the complexation ability of the calixarene, and there exist calixarene–alkanethiol mixed monolayer compositions in which the particles become unstable due to mismatching ligand spacer lengths, (iii) calixarenes with very short spacers bound to the gold surface can experience an enhancement in the delocalized π-electron density available for cation complexation, likely due to the proximity of the gold-bound sulphur to the calixarene cavity.
Introduction
Self-assembly is an event that occurs when a force originating from local interactions drives compounds of different chemistries together to form larger structures. Materials exhibiting self-assembly properties hold great promise as they are likely to play a crucial role in nanomaterial preparation techniques, which in turn may achieve unparallelled molecular precision in the material preparation of the future. It is clear that self-assembly lies at the core of both synthetic nanomaterial creation and the organization of such materials, and thus in the core of the modern paradigm of Nanoarchitectonics.1Calixarene-based host–guest sensing on gold surfaces has recently received a lot of attention from the scientific community, most likely because of the diverse chemical modification possibilities of the calixarene2 and the well-established synthetic preparation methods of the gold nanoparticles.3,4 Many groups have extensively studied calixarene–gold hybrids for various purposes, in which the integral themes often are supramolecular structures via host–guest interactions,5,6 molecular recognition and sensing,7 or metal surface accessibility control,8 all of which are strongly reliant of self-assembly behavior.
The properties of AuNPs are dictated by nanoparticle size (quantum size effect: the properties of matter scales with the particle diameter) and protecting agent (material bound to particle surface confers nanoparticle stability, solubility and chemical properties.) This duality of the properties of particles indicates the importance of mixed monolayers on nanoparticle surfaces: they allow additional properties to the material or – as in this study – the possibility of diluting the amount of active component (calixarenes) on the nanoparticle surface. It is clear that the self-assembly properties of the material can be tuned by altering the concentration of the active self-assembling compound on the AuNP surface.
In a previous publication, our group investigated the correlation between nanoparticle-bound calixarene host–guest complexation and the aggregation of the NPs.5 One of the preliminary observations was that the addition of alkanethiols of certain length can fine-tune the complexation ability of the surface-bound calixarene. The observation provoked the question how the self-assembly behavior would work in reversed conditions, that is, in a scenario where only a few calixarenes stand on the nanoparticle surface among a large excess of surrounding alkanethiols. So far, detailed studies on the complexation ability of calixarene compounds in such a mixed monolayer attached to a gold surface remains largely untouched in the literature. Although some work has already been done,9 there is certainly room for a more detailed study.
The ability of fine-tuning the activities of receptors on AuNPs is an important tool controlling the nanoparticle self-assembly into larger supramolecular structures. In this study we examine the complexation ability of calixarenes with different aliphatic spacers under different conditions: (i) in a mixed monolayer of alkanethiols and calixarenes (less than 11 mol% of calixarenes) on AuNP surfaces, (ii) in pure calixarene monolayer (100 mol%) on AuNPs, and (iii) in free calixarenes in solution. In the mixed monolayer case, special focus was on how the lengths of the aliphatic chains of the calixarene and the alkanethiol affect the calixarene complexation ability. The guest molecule was chosen to be cetyl pyridinium chloride (Pyr-C16), as in the previous study. Interesting data on calixarenes diluted in mixed monolayers on gold, and on calixarene cavity situated in a very close proximity of the gold surface is obtained. It is also presented how the simple combination of NMR, TEM and TGA analysis methods can be employed to obtain very detailed information on AuNP monolayer composition and coverage, a topic widely studied at present.10
Materials and experimental
Cetyl pyridinium chloride (Pyr-C16), dibromobutane (DBB), butanethiol (BT), dodecanethiol (DT), tetraoctyl ammonium bromide (TOAB) and other chemicals were purchased from Aldrich or Fluka. Solvents were distilled and reactants dried in a vacuum desiccator prior to use. 1-Tosyloxyundec-10-ene was synthesised according to existing method.11 The Calix0 and Calix2 were synthesised as described elsewhere in the literature.5,6,9,11 The calixarenes referred to in this paper are denoted by aromatic hydroxyl substitution number (Calix0, Calix2, Calix4) and their structures are shown in Fig. 1 and 2. | ||
| Fig. 1 Synthesis protocol for Calix2. | ||
 | ||
| Fig. 2 Synthesis protocol for Calix4. | ||
Synthesis of Calix2
Calix2 was synthesized as described in the literature.9,11 Briefly, Calix0 (12.4 mmol) was mixed with K2CO3 (124.0 mmol) in acetonitrile (170 ml). 1-Tosyloxyundec-10-ene (24.8 mmol) was added after stirring the mixture for 1 h at room temperature. The reaction was run for 3 days under reflux conditions. The reaction mixture was concentrated in a vacuum desiccator, dissolved in dichloromethane and then extracted with 1 M HCl and water. The product was dried with Na2SO4 and evaporated to dryness. The crude product was purified using column chromatography (SiO2, CH2Cl–cyclohexane 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, Rf 75%). Yield was 38%. NMR showed full substitution of the two aromatic hydroxyl groups per calixarene. 1H NMR (300 MHz, CDCl3) 8.25 (2H, s), 7.07 (4H, d), 6.93 (4H, d), 6.80–6.63 (4H, m), 5.82 (2H, m), 4.98 (4H, m), 4.33 (4H, d), 4.01 (4H, t), 3.38 (4H, d), 2.07 (4H, m), 1.71 (4H, m), 1.5–1.3 (24H, m).
:25, Rf 75%). Yield was 38%. NMR showed full substitution of the two aromatic hydroxyl groups per calixarene. 1H NMR (300 MHz, CDCl3) 8.25 (2H, s), 7.07 (4H, d), 6.93 (4H, d), 6.80–6.63 (4H, m), 5.82 (2H, m), 4.98 (4H, m), 4.33 (4H, d), 4.01 (4H, t), 3.38 (4H, d), 2.07 (4H, m), 1.71 (4H, m), 1.5–1.3 (24H, m).
Product of the previous reaction (4.7 mmol) was dissolved in toluene (120 ml) with thioacetic acid (18.6 mmol) under argon flow. After 30 min of stirring and argon bubbling, a catalytic amount of AIBN was added and the mixture was refluxed. NMR was used to monitor the disappearance of the double bond: if the signals persisted, the reaction mixture was cooled to room temperature and more AIBN was added and the reflux was continued. Once the reaction was completed, the solvent was evaporated and the crude product was dissolved in CH2Cl2 and extracted with a concentrated NaHCO3 solution and water, and then dried over Na2SO4. The product was thoroughly dried and dispersed in ethanol (70 ml, poor solubility). Concentrated MeOH–MeONa solution was added under vigorous stirring and the mixture was further stirred at room temperature for 24 h. Solvent was evaporated, the solid residue was dissolved in CH2Cl2 and extracted with 2 M HCl and water (emulsifies very easily). After Na2SO4 drying and thorough drying in vacuum, the product was recrystallized from chloroform–MeOH mixture. Yield: 59%. 1H NMR (500 MHz, CDCl3) 8.24 (2H, s), 7.06 (4H, d), 6.93 (4H, d), 6.80–6.62 (4H, m), 4.34 (4H, d), 4.01 (4H, t), 3.39 (4H, d), 2.70 (4H, t), 2.10 (4H, m) 1.71–1.28 (34H, m).
Synthesis of Calix4
Calix4 was synthesised as reported.12 The synthesis protocol, briefly: dry DMF (190 ml) was placed into a flask and purged with nitrogen. Calix0 (9.4 mmol), dibromobutane (188.5 mmol) and NaH (56.6 mmol) were added into the flask. (Caution: NaH reacts violently with water!) The mixture was stirred for 20 min, after which the flask was heated to 80 °C. The reaction mixture was stirred under N2 for 5 days.The reaction was quenched with careful water addition and the mixture was extracted with chloroform twice. The organic phase was washed with distilled water twice and dried with anhydrous sodium sulphate. Organic phase was evaporated under high vacuum (3 mbar, 140 °C) in order to remove volatile organic substances and the most of the DBB. The residue was purified using column chromatography (chloroform–hexane 75:25, Rf 80%.). The yield was 49%. 1H NMR (300 MHz, CDCl3) 6.3 (12H, s), 4.1 (4H, d), 3.6 (8H, t), 3.2 (8H, t), 2.9 (4H, d), 1.7 (16H, m).
The product (3.8 mmol) of previous synthesis phase was placed in a nitrogen purged flask containing 125 ml of dry DMF. Thiourea (41.5 mmol) was added and the mixture was stirred for 20 min, after which the flask was heated to 80 °C. The mixture was stirred under nitrogen for 12 hours and then, was quenched by pouring the mixture into NaOH solution (3.8%, 580 ml). The reaction mixture was stirred for 1 hour and finally the pH was adjusted to 4–5 using HCl.
Product was filtered, washed with water, dried in vacuum and further purified using column chromatography (chloroform, Rf 75%.). Yield: 70%. 1H NMR (300 MHz, CDCl3) 6.6 (12H, s), 4.4 (4H, d), 3.9 (8H, t), 3.2 (4H, d), 2.6 (8H, q), 2.0 (8H, m) 1.7 (8H, m), 1.4 (4H, t).
Synthesis of gold nanoparticles
Calix2–AuNP, DT/Calix2–AuNP, Calix4–AuNP and BT/Calix4–AuNP were synthesised using a modified version of the general method present in the literature.5,13 Briefly, 1.18 mmol of HAuCl4·x3H2O was dissolved in 10 ml of distilled water. TOAB (4.12 mmol) was added into 90 ml of toluene. The solutions were mixed and stirred at room temperature until all the gold had transferred to the organic phase. Excess water was removed using a Pasteur pipette. The solution was stirred vigorously and freshly prepared NaBH4 (16.48 mmol) in 10 ml water was injected at a rate roughly of 1 ml per second into the reaction flask. Mixture was stirred at high speed for 2 h.The toluene phase was washed in an extraction funnel to remove NaBH4 residues: once with 0.1 M H2SO4 (150 ml), twice with 1 M Na2CO3 (2 × 75 ml) and three times with distilled water. The organic phase was dried over anhydrous MgSO4 and the toluene liquid was filtered.
This stock nanoparticle solution was divided into separate flasks and a calculated feed amount of ligand mixtures were added into each of the flasks: DT–Calix2 mixture 2% in calixarene, two BT–Calix4 mixtures 2% and 5% in calixarene, pure Calix2 and pure Calix4 nanoparticles (100% calixarene content for both). The ligand exchange reactions were stirred for one week at room temperature.
Particles were precipitated with ethanol, poured on a glass filtration frit (porosity type 5) and large amount of ethanol and acetone were poured through the nanoparticles immobilized on the frit. The nanoparticles were collected from the frit with minimal amount of chloroform. To each 15 ml of chloroform, 25 ml of MeOH was added. Solution was centrifuged at 5000 rpm (3773 RCF) for 15 min. The supernatant was eliminated and the precipitate taken up with 15 ml chloroform. This solution was centrifuged at 1500 rpm (340 RCF) for 10 min. Supernatant was collected and diluted with 40 ml of MeOH and centrifuged at 10000 rpm (8720 RCF) for 20 min. Supernatant was removed and the precipitate was dispersed in minimal amount of chloroform. The particles were kept in solution in a freezer for months without visible aggregation. The yields were, in gold, 48% for Calix2–AuNP, 50% for DT/Calix2–AuNP, 31% for Calix4–AuNP, and 32% for BT/Calix4–AuNP.
Synthesis of BT/Calix2–AuNP
BT/Calix2–AuNP could not be prepared in a manner described above: both direct Brust–Schiffrin (adding NaBH4 into a mixture of ligands, solvent and gold salt) and Brust–Schiffrin ligand exchange (the method used for the other particles) resulted in an insoluble precipitate during the purification phase. Finally, particles were successfully prepared by synthesising first pure BT–AuNPs and then ligand exchanging Calix2 onto the particles. Detailed synthesis protocol in brief:HAuCl4·x3H2O (0.88 mmol) was dissolved in 10 ml of deionized water. TOAB (2.21 mmol) was dissolved in 70 ml of toluene. These solutions were mixed and stirred until the gold ions had transferred to the organic phase, after which the excess water was removed using a pipette. Butanethiol (0.88 mmol) was added to the solution and the reaction mixture was stirred for 10 minutes. Freshly prepared NaBH4 water solution (8.83 mmol, 10 ml) was injected to the reaction mixture at a rate of roughly 1 ml per second and the mixture was stirred vigorously for 3.5 hours at room temperature.
The dark organic phase was collected, solvent concentrated with rotary evaporator and the product was precipitated using ethanol. The precipitate was poured on a glass filtration frit (porosity 5) and washed with at least 80 ml of ethanol and 150 ml of acetone. Particles were collected from the frit by using a minimal amount of toluene. Particles were analysed using TGA (mass loss of 15.4%).
Using the TGA data, a Calix2 amount matching 2 mol% of organics present on the nanoparticles was calculated. The corresponding amount of Calix2 was added to the toluene solution. Reaction mixture was stirred at room temperature for one week to allow maximal ligand exchange. The particles were purified by precipitating them with ethanol, pouring them on a glass frit and washing them with large amount of ethanol and acetone. Particles were collected from the frit using minimum amount of chloroform. Yield: 78% in gold.
Characterization
Thermogravimetry (TGA) measurements were done under a flowing nitrogen atmosphere using Mettler-Toledo TGA850 equipment with STARe software. The temperature range was 25–800 °C and a heating rate of 10 °C min−1 was used. TGA results were used to estimate the amount of calixarene on the particles. This data (alongside the NMR monolayer composition data) was used to calculate the calixarene and guest amounts to be used in the NMR complexation studies.For transmission electron microscopy (TEM), diluted dispersions of nanoparticles (∼1 mg ml−1) were dried onto a holey carbon grid (copper mesh), which were then observed using bright-field TEM on a Tecnai 12 transmission electron microscope (operating voltage 120 kV). Images were analyzed with the ImageJ software.14 Several hundreds of nanoparticles per sample were included in the size distribution determination.
NMR studies for reaction products were performed on a Bruker 500 MHz NMR system. The mixed monolayers were detached from the surface by using the Iodine Death reaction.15,16 Briefly, iodine was added to the NMR tube containing the nanoparticles and the tube was shaken for several minutes. After allowing the tube to stand for a few minutes, NMR spectrum was recorded. It was observed that the broad signals arising from surface-bound nanoparticles were now replaced with sharp signals originating from the free ligands. All of the proton NMR measurements were done in deuterated chloroform, 128 pulses, 5 s relaxation delays. Using the TGA and NMR/Iodine Death data obtained, the amount of calixarene per each nanoparticle was determined. An accurately weighted amount of nanoparticles was dispersed into 1 ml of deuterated chloroform. A concentrated solution of the guest (Pyr-C16) was prepared separately by accurately measuring a known amount of the substance and dissolving it into a known volume of deuterated chloroform. The amount of the guest solution required to achieve a desired calixarene:guest ratio was calculated using the TGA/NMR data. A required amount of the guest solution was added into the AuNP NMR sample tube using a micropipette. The tube was shaken for a few minutes to mix the contents. After measuring the NMR spectrum for one ratio, another calculated dose of the guest solution was added into the same NMR tube. This cycle was repeated until the NMR spectra were recorded for all calixarene:guest ratios ranging from 6.0 to 0.25 or 0.07. NMR spectra of the aromatic areas of pyridinium and calixarene were of particular interest, because these signals are known to shift positions upon complexation,17 due to the alterations in the electronic environment of the guest molecules when they enter the calixarene cavity.
Results and discussion
Nanoparticle synthesis and characterization
A series of AuNPs with mixed monolayers were prepared. The protecting monolayers on the particles were mixtures of alkanethiols (butanethiol (BT) or dodecanethiol (DT)) and calixarenes. Calixarenes were substituted with either two C11 or four C4 alkanethiol spacers. Particles coated with only calixarenes were prepared, as well.Nanoparticles were fully characterized using NMR (mixed monolayer composition), TGA (amount of organic compounds) and TEM (nanoparticle size). A typical micrograph and particle size distribution are shown in Fig. 3 and a summary of the results is given in Table 1.
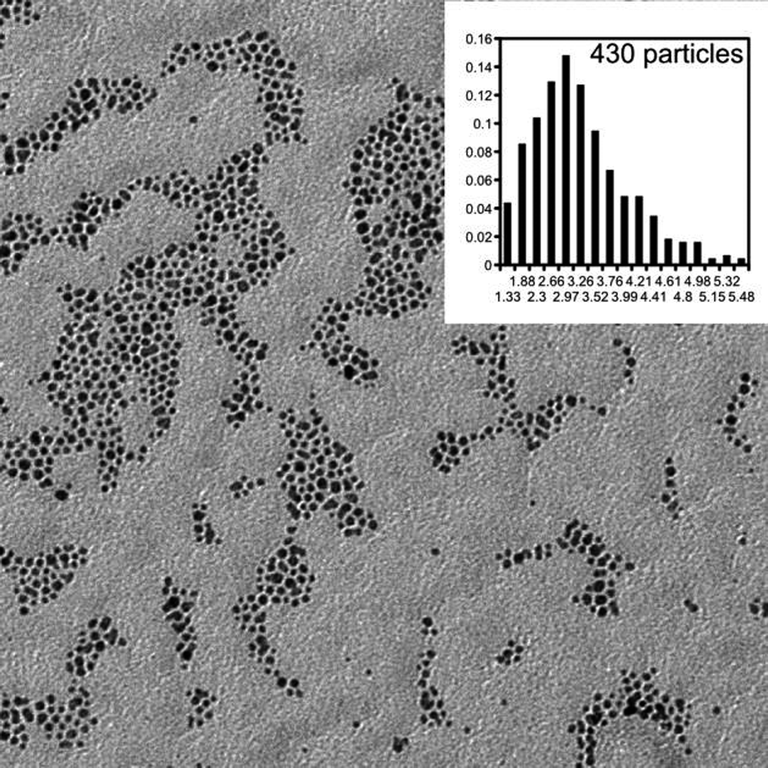 | ||
| Fig. 3 TEM micrograph of BT/Calix4–AuNP. Size distribution is shown as inset. | ||
| Sample | Diameter/nm | Calixarene in feed/mol % | Calixarene on particle/mol % | Average structure | Ligand footprint/Å2 |
|---|---|---|---|---|---|
| a From ref. 5.b Synthesised using a different method, see Experimental section.c Maximum calixarene amount.d Minimum footprint.e From ref. 18. | |||||
| Calix2–AuNpa | 3.1 ± 0.7 | 100 | 100 | Au635Calix57 | 45.8 |
| DT/Calix2–AuNP | 4.0 ± 1.0 | 2 | 4.7 | Au1357DT212Calix10 | 19.3 |
| BT/Calix2–AuNPb | 1.9 ± 0.4 | 2 | 0.8 | Au154BT52Calix0.5 | 19.4 |
| Calix4–AuNp | 3.1 ± 0.6 | 100 | 100 | Au605Calix37c | 66.9d |
| BT/Calix4–AuNp | 3.1 ± 0.8 | 5 | 10.7 | Au647BT66Calix8 | 35.3 |
| BT/Calix4–AuNp | 3.1 ± 0.5 | 2 | 3.5 | Au654BT123Calix5 | 20.7 |
| DT–AuNPe | 3.0 ± 1.8 | 0 | 0 | Au570DT110 | 22 |
It should be noted that higher amount of Calix2 on the BT/Calix2–AuNP resulted in energetically disfavored conditions that adversely affect nanoparticle stability. Particles with very low amount of Calix2 (0.8 mol%) were stable. Any attempt to increase Calix2 content by mild heating (35 °C) in an extended ligand exchange resulted in total insolubility of the nanoparticles after the particles were precipitated with EtOH, i.e. the particles did not redisperse in any medium. DT/Calix2–AuNP (Calix2 content 4.7 mol%) and BT/Calix4–AuNP (Calix4 content 3.5 and 10.7 mol%), on the other hand, formed stable dispersions, which did not precipitate during several months of storage in the freezer. The similarity between these particles is that the aliphatic spacers of the ligands are of similar size (12 carbon dodecanethiol vs. 11 carbon Calix2 on one hand, and 4 carbon butanethiol vs. 4 carbon Calix4 aliphatic chains, on the other). BT/Calix2–AuNP contained a mixed monolayer of 4 carbon butanethiol and 11 carbon Calix2 aliphatic chains. This sample, in contrast to the other ones, precipitated immediately upon purification, and could be prepared only via a different synthesis route with very low Calix2 amount (0.8 mol%). Attempts to increase Calix2 content resulted in unstable particles.
It is clear that there exists some kind of mixed monolayer composition that destabilizes the nanoparticles. We attribute this instability to the mismatch in protecting agent spacer dimensions, i.e. the butanethiol C4 chain and Calix2 C11 spacer, which may promote particle aggregation, possibly via an interdigitation mechanism.5,19,20
We were able to synthesize a batch of stable mixed monolayer BT/Calix2 nanoparticles, but only with very low Calix2 amount and a different particle diameter than the other nanoparticles described herein, see Table 1. The BT/Calix2–AuNPs seemed to be stable when stored in dispersion in a freezer, but keeping the sample at room temperature for a week resulted in permanent precipitation, showing their unstable nature. The instability of BT/Calix2–AuNP was in contrast to BT/Calix4–AuNP, Calix4–AuNP, Calix2–AuNP and DT/Calix2–AuNP, which were stable for months.
NMR spectroscopy was employed on AuNPs before and after the Iodine Death reaction (ESI S1 and S2†). A typical result is shown in Fig. 4, spectra (b) and (c). The sample without iodine gives very broad aromatic signals at the same position as the free ligand. This indicates that the particles are free of calixarene that is not attached to the surface. Murray et al. have previously shown that the NMR signals originating from different parts of alkanethiol chains bound to gold surface exhibit different signal broadening: the closer the atoms are to the gold surface, the broader NMR signals they produce.20 In this context, one might expect to observe sharp aromatic signals from calixarenes bound to the gold surface. However, our data clearly shows that surface bound aromatic signals are significantly broadened into the baseline of the NMR spectrum, regardless of the length of the calixarene aliphatic spacer. The observed severe broadening is attributed to rigidity of the aromatic ring system of the calixarenes, which, moreover, are sterically locked and bound to the gold surface via multiple thiolate bonds. This should result in a more solid-like, motionally suppressed organic system with fast spin relaxations,21 when compared to flexible alkanethiols on a gold surface.
 | ||
| Fig. 4 NMR spectra of (a) free Calix4, (b) BT–Calix4–AuNP before Iodine Death, (c) BT–Calix4–AuNP after Iodine Death. | ||
Upon iodine addition the protecting ligands are detached quantitatively15 from the nanoparticle surface, resulting in the appearance of sharp calixarene signals. This enables the reliable determination of the mixed monolayer molar composition by simply integrating the aromatic region (single peak at ∼6.6 ppm for Calix4 and four peaks at ∼6.6–7.1 ppm for Calix2) and the aliphatic proton region (∼2.7 ppm, –CH2–SH groups from both calixarene and alkanethiols.) The signals are marked with arrows in spectrum 4c. The additional peaks observed in 4c are likely due to a mixture of disulfides which build up upon the detachment of the thiols. For particles prepared with Brust–Schiffrin reaction, some tetraoctyl ammonium bromide residues are also known to appear in NMR after the Iodine Death reaction.16
All the gold-bound thiol compounds showed the same NMR signal broadening, except Calix4–AuNP, see ESI S3.† This nanoparticle produces visible NMR signals from the surface bound calixarene even without the Iodine Death reaction, which is in contrast to the other nanoparticles described here. The signals in Calix4–AuNP are attributed to free Calix4 disulfide contamination in the sample, some of which remained despite of the extensive purification.
Calix2 and Calix4 have very similar molar masses, and in the nanoparticles Calix2–AuNP and Calix4–AuNP, also the gold cores are identical in size. Since Calix2–AuNP and Calix4–AuNP show similar mass losses in TGA (see ESI S4†), one can conclude the amount of free disulfide contamination is not very significant.
Assuming that the single thiolate ligand footprint is close to 22 Å2 (literature value for dodecanethiol protected AuNPs22), one could estimate from Table 1 that in Calix2–AuNP, the calixarene stands with both of its two thiol groups on the particle surface (ligand footprint for Calix2–AuNP is 45.8 Å2). It should be noted, however, that in Table 1, the calixarene amount for Calix4–AuNP can be described only as a maximum amount, and the footprint as a minimum footprint, because of the presence of the free Calix4 disulfide contaminant. For Calix4–AuNP, the data suggests that the calixarene is in fact – on average – attached to the surface with three or all four thiol groups (ligand footprint minimum of 66.9 Å2).
However, it should be also mentioned that our ligand footprint determination methods (combining of TEM, NMR and TGA data) can only obtain the average ligand footprint. The possibility of mixture of Calix4s attached with three or four legs cannot thus be completely ruled out. A detailed study, which elucidates Calix4 footing on more accurately measurable gold nanoclusters has been published elsewhere.23
In Table 1, the mixed monolayers of alkanethiols and calixarenes show footprints close to that of dodecanethiol of gold surface (22 Å2). Due to the high number of alkanethiols in the samples, the average footprint should approach the footprint of the dodecanethiol.
An interesting common denominator seen in Table 1 is the relation of calixarene feed in the reaction and the obtained calixarene composition in the mixed monolayer: approximately twice as much calixarene was observed on the nanoparticles when compared to the amount put into the synthesis feed. This is attributed to the stronger surface binding of the multi-thiolate calixarenes compared to single-thiolate alkanethiols: during the ligand exchange of TOAB into an excess of a calixarene–alkanethiolate mixture, stronger calixarene binding prevails. This is not the case for BT/Calix2–AuNP, because this nanoparticle could only be prepared by first synthesising pure BT–AuNP and then ligand exchanging Calix2 onto the surface, i.e. the surface was pre-covered with a strongly bound alkanethiolate monolayer, inhibiting the penetration and binding of the Calix2 to the gold surface.
Complexation experiments
Complexation of cetyl pyridinium chloride guest (Pyr-C16) with calixarene on AuNPs has been studied by NMR. Results of the complexation experiments are shown in the ESI (S5–S11†) and are summarized in Fig. 5 and tabulated in Table 2. The position of various chemical shifts were monitored in an NMR experiment where the calixarene:pyridinium ratio was changed by titration with pyridinium. In Fig. 5 the data is expressed as a change in the chemical shift from pure pyridinium signals. Upon saturation of the calixarene with the pyridinium, a distinct change of slope is observed. The inflection points are expressed in Table 2 as the Calixarene Saturation Ratio (CSR), which provides information about the calixarene accessibility. The origin of the change in chemical shifts is the change in the chemical environment of the pyridinium as it enters the calixarene cavity,17 and the upfield shift indicates more shielded pyridinium protons. The change in the chemical shift is a measure of the amount of electron density of the calixarene host that interacts with the pyridinium guest.
 | ||
| Fig. 5 Pyridinium NMR signal position shift between free pyridinium and varying calixarene:pyridinium ratios. (a) Free Calix2, (b) Calix2–AuNP, (c) BT/Calix2–AuNP, (d) DT/Calix2–AuNP, (e) free Calix4, (f) Calix4–AuNP, (g) BT/Calix4–AuNP. The inset shows the monitored pyridinium signals. | ||
:pyridinium ratio, the complexation experiment inflection point
| Sample | Type | CSR | Max delta γ |
|---|---|---|---|
| a From ref. 5.b Measured from pyridinium ε-peak shifts, because other datasets were not complete due to excessive peak shifting (pyridinium peaks overlapped with other peaks invalidating reliable evaluation). See Fig. 5a.c Maximum pyridinium γ-peak could not be determined due to peak overlapping.d No observable inflection point, see Fig. 5g. | |||
| Free Calix2 | Calix2 | 0.81b | Highc |
| Calix2–AuNPa | Au635Calix57 | 0.83b | 0.77 |
| BT/Calix2–AuNP | Au154BT52Calix0.5 | 0.31 | 0.27 |
| DT/Calix2–AuNP | Au1357DT212Calix10 | 0.22 | 0.36 |
| Free Calix4 | Calix4 | 0.52 | 0.13 |
| Calix4–AuNP | Au605Calix37 | 0.52 | 0.29 |
| BT/Calix4–AuNP | Au647BT66Calix8 | —d | 0.09 |
Essentially, Table 2 shows, that the calixarene accessibility remains the same when comparing free calixarene in solution and the calixarene bound to AuNP surface: the CSR values are the same for free Calix2 and Calix2–AuNP. Addition of alkanethiols decrease the accessibility, depending on the alkanethiol chain length: the lowest accessibility is obtained for DT. Similar behavior is observed for free Calix4 and Calix4–AuNP, and the addition of short alkanethiol (BT/Calix4–AuNP) makes the Calix4 completely inaccessible for the pyridinium. Since the calixarene is completely inaccessible in BT/Calix4–AuNP, but remains accessible in BT/Calix2–AuNP, one can conclude that the aliphatic chain length compared to calixarene spacer length is a critical factor to take into account when synthesising a mixed monolayer of tuneable receptor activity.
The electron density in the calixarene available for the pyridinium (max delta γ in Table 2) changes depending on the monolayer composition. For Calix2 family, the max delta γ decreases following the trend: free Calix2 > Calix2–AuNP > DT/Calix2–AuNP. It is important to note that BT/Calix2–AuNP has not been included in this comparison, because its size (1.9 nm) is different from the other nanoparticles (∼3 nm): in the nanoscale, the particle size may affect the material properties in an unexpected way24,25 and the effects seen may very well be due to the different particle sizes.
When comparing nanoparticles in the same size regime, the Calix4 family shows a different max delta γ trend than the Calix2 family: Calix4–AuNP > free Calix4 > BT/Calix4–AuNP. The data suggests that the π electron density available for complexation in the calixarene cavity increases when Calix4 binds to gold surface. Furthermore, the shapes of the complexation curves 5e and 5f clearly indicate that the complexation behavior of the species are different. The delta ppm is negative for the epsilon protons of the pyridinium in case of free Calix4 in solution.
It is known in the literature that the gold atoms lose d-electrons when alkanethiols are attached to the surface,26 and that charge transfer occurs from the NP to the thiol.27 There are also reports of electron-rich calixarene cavities directly interacting with the electron-depleted gold surface, when the spacer is short enough.12
In this light, the enhanced change of the electronic environment (max delta γ) of the gold-bound Calix4 when compared to free Calix4 can be attributed to the proximity of negatively charged surface-bound sulphur species to the calixarene cavity. As stated previously, the sample does contain free Calix4 as an impurity, but its effect should be negligible due to its low amount. Also, the presence of free Calix4 impurity should decrease, whereas the surface binding of the Calix4 seems to increase max delta γ, as seen from Table 2. It is clear from the data that BT/Calix4–AuNP has no complexation ability. This indicates that the butanethiol of similar length as the Calix4 aliphatic C4 spacer blocks Pyr-C16 access to the calixarene cavity. Table 2 shows similar, but weaker effect for DT/Calix2–AuNP: the Calixarene Saturation Ratio is lower for DT/Calix2–AuNP than for Calix2–AuNP and BT/Calix2–AuNP. The addition of DT does not completely inhibit the compexation ability of Calix2, whereas the addition of BT inhibits Calix4. This is most likely because of Calix2 has, per se, higher complexation ability due to the presence of free hydroxyl groups.28
Conclusions
Combination of calixarenes and alkanethiols in a protecting monolayer on AuNPs is a method to adjust the host–guest complexation of the calixarenes. The calixarene receptors remain active even with a very low quantity of the calixarenes diluted among alkanethiols. The relative sizes of the alkanethiols and the spacers between the macrocycles and gold surface are the key parameters controlling the complexation of the calixarene. It has been observed that with certain monolayer compositions the particles may turn colloidally unstable. When the calixarenes are bound very close to the metal surface with a short spacer, the π-electron density in the cavity increases which affects the complex formation with the cation examined in this study. This probably owes to the proximity of the electronegative surface-bound sulfur to the calixarene cavity. The results obtained in this study indicate that the synthesis of materials with controllable and tuneable mixed monolayers of calixarenes and alkanethiols on the gold surface can offer exciting possibilities for future self-assembly systems.Acknowledgements
We would like to thank the FUNMAT Center of Excellence for Functional Materials and the Academy of Finland (project no. 127329 and project no. 256314) for financial support. This work made use of the Aalto University Nanomicroscopy Center premises.References
- P. Kujawa and F. M. Winnik, Langmuir, 2013, 29, 7354–7361 CrossRef CAS PubMed.
- C. D. Gutsche, Calixarenes, RSC, 1989 Search PubMed.
- Y. Li, O. Zaluzhna, B. Xu, Y. Gao, J. M. Modest and Y. J. Tong, J. Am. Chem. Soc., 2011, 133, 2092–2095 CrossRef CAS PubMed.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- P. M. S. Pulkkinen, S. Wiktorowicz, V. Aseyev and H. Tenhu, RSC Adv., 2013, 3, 733–742 RSC.
- F. Ciesa, A. Plech, C. Mattioli, L. Pescatori, A. Arduini, A. Pochini, F. Rossi and A. Secchi, J. Phys. Chem. C, 2010, 114, 13601–13607 CAS.
- H. J. Kim, M. H. Lee, L. Mutihac, J. Vicens and J. S. Kim, Chem. Soc. Rev., 2012, 41, 1173–1190 RSC.
- J.-M. Ha, A. Solovyov and A. Katz, Langmuir, 2009, 25, 10548–10553 CrossRef CAS PubMed.
- A. Arduini, D. Demuru, A. Pochini and A. Secchi, Chem. Commun., 2005, 645–647 RSC.
- H. Hinterwirth, S. Kappel, T. Waitz, T. Prohaska, W. Lindner and M. Lämmerhofer, ACS Nano, 2013, 7, 1129–1136 CrossRef CAS PubMed.
- R. Métivier, I. Leray, B. Lebeau and B. Valeur, J. Mater. Chem., 2005, 15, 2965 RSC.
- J.-M. Ha, A. Katz, A. B. Drapailo and V. I. Kalchenko, J. Phys. Chem. C, 2009, 113, 1137–1142 CAS.
- M. Brust, D. Bethell, C. J. Kiely and D. J. Schiffrin, Langmuir, 1998, 14, 5425–5429 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- A. C. Templeton, M. J. Hostetler, C. T. Kraft and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 1906–1911 CrossRef CAS.
- Z. Tang, D. A. Robinson, N. Bokossa, B. Xu, S. Wang and G. Wang, J. Am. Chem. Soc., 2011, 133, 16037–16044 CrossRef CAS PubMed.
- S. Ishihara and S. Takeoka, Tetrahedron Lett., 2006, 47, 181–184 CrossRef CAS PubMed.
- A. Määttänen, P. Ihalainen, P. Pulkkinen, S. Wang, H. Tenhu and J. Peltonen, ACS Appl. Mater. Interfaces, 2012, 4, 955–964 Search PubMed.
- A. Swami, A. Kumar and M. Sastry, Langmuir, 2003, 19, 1168–1172 CrossRef CAS.
- R. H. Terrill, T. A. Postlethwaite, C. Chen, C.-D. Poon, A. Terzis, A. Chen, J. E. Hutchison, M. R. Clark and G. Wignall, J. Am. Chem. Soc., 1995, 117, 12537–12548 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- R. G. Shimmin, A. B. Schoch and P. V. Braun, Langmuir, 2004, 20, 5613–5620 CrossRef CAS.
- J. Hassinen, P. Pulkkinen, E. Kalenius, T. Pradeep, H. Tenhu, H. Häkkinen and R. H. A. Ras, J. Phys. Chem. Lett., 2014, 585–589 CrossRef CAS.
- W. H. Qi, Phys. B, 2005, 368, 46–50 CrossRef CAS PubMed.
- G. Ozin and A. Arsenault, Nanochemistry: A chemical approach to nanomaterials, The Royal Society of Chemistry, 2005 Search PubMed.
- P. Zhang and T. K. Sham, Appl. Phys. Lett., 2002, 81, 736 CrossRef CAS PubMed.
- P. Zhang and T. K. Sham, Phys. Rev. Lett., 2003, 90, 245502 CrossRef.
- L. J. Bauer and C. D. Gutsche, J. Am. Chem. Soc., 1985, 107, 6063–6069 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00494a |
| This journal is © The Royal Society of Chemistry 2014 |