Transition metal-free iodine-promoted haloamination of unfunctionalized olefins†
Wei Li,
Gong-Qing Liu,
Bin Cui,
Li Zhang,
Ting-Ting Li,
Lin Li,
Lili Duan and
Yue-Ming Li*
College of Pharmacy and Tianjin Key Laboratory of Molecular Drug Research, Nankai University, 94 Weijin Road, Tianjin 300071, People's Republic of China. E-mail: ymli@nankai.edu.cn; Fax: +86 22 23507760; Tel: +86 22 23504028
First published on 4th March 2014
Abstract
A transition metal-free route to 3-halopiperidines and 2-halomethylpiperidines was described. In the presence of iodine, potassium persulfate and a suitable halogen source, intramolecular haloamination of 4-penten-1-amines and 5-hexen-1-amines proceeded readily, leading to the corresponding substituted piperidines in good isolated yields.
Vicinal haloamines such as 3-halopiperidines or 2-halomethylpiperidines have been used as important subunits in mechanism-based anti-tumour compounds,1 they also constitute important parts in several natural products.2
Among the variety of methods developed, direct intramolecular haloamination of unfunctionalized 4-penten-1-amines or 5-hexen-1-amines would be one of the most straightforward routes to heterocyclic vicinal haloamines such as 3-halopiperidines or 2-halomethylpiperidines. The first generation direct haloamination of 4-penten-1-amines involved the utilization of dihalogens, and generally suffered from functional group tolerance due to the high reactivity of dihalogen.3 The second generation chloroamination of 4-penten-1-amines developed by Göttlich et al. involved a CuI-catalyzed free radical cyclization of a pre-functionalized N–Cl substrate.4 The reactions were carried out at elevated temperature, and 3-chloropiperidine products were obtained in good yields. A Lewis acid–TiCl3 system was also reported to promote the free radical cyclization of N–Cl substrates at low temperature.5 Again, functionalization of the substrates to the corresponding N–Cl derivatives was required prior to the cyclization. Later, Chemler,6 Lu,7 Michael,8 Muñiz9 and Liu10 realized the intramolecular haloamination of N-sulfonamides, amides or N-carbamates using PdII-catalyzed cyclizations, and the corresponding 2-halomethyl heterocycles or 3-halopiperidines were obtained in good to excellent yields. In these reactions, palladium was used to activate the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, subsequent aminopalladation produced a C–Pd intermediate which could be cleaved by an additional molecule of copper halide to produce the final haloamination products. Recently, hypervalent iodines were also used in aminocyclization of several unfunctionalized olefins, but additional additives would generally be needed to complete the reactions.11
C double bonds, subsequent aminopalladation produced a C–Pd intermediate which could be cleaved by an additional molecule of copper halide to produce the final haloamination products. Recently, hypervalent iodines were also used in aminocyclization of several unfunctionalized olefins, but additional additives would generally be needed to complete the reactions.11
In addition to these methods, electrophile-induced cyclization of N-substituted alkenylimines developed by De Kimpe et al. was also one of the most efficient methods for the production of functionalized pyrrolidines and/or piperidines. For example, intramolecular cyclizations of N-substituted iminium olefins with PhSeX produced selenyl substituted cyclic iminium compounds which could be conveniently converted to the corresponding pyrrolidines or piperidines upon reductive deselenylation.12 Cyclization of γ,δ-alkyenylimines with bromine produced the cyclic iminium bromides in good yields, and reduction of the thus obtained cyclic iminium bromides with sodium borohydride provided the corresponding bromopyrrolidines in excellent yields.13 Pyrrolidines could also be obtained upon free radical debromination with Bu3SnH–AIBN.14 Cyclization of γ,δ-unsaturated aldimines with bromine produced the corresponding cyclic iminium bromides, and 5-alkoxy-1,2,3,4-tetrahydropyridines could be obtained via thermal rearrangement of 2,5-dialkoxypiperidines.15 Starting from alkenylsulfinimines or alkenylsulfinamides, several functionalized cyclization products could also be obtained via electrophile-induced cyclization using phenylselenyl bromide, iodine and bromine as nucleophiles. Subsequent functional group transformation furnished a variety of functionalized pyrrolidines and piperidines.16 Hydride-promoted ring expansion of 2-azaspiropyrrolinium salts produced the N-heterocyclic compounds, and alkaloids such as (−)-nitramine could be easily prepared in good overall yield using this method.17
We have shown that CuCl2 alone could act as both reaction promoter and the chlorine source, and Markovnikov vicinal chloroamines such as 2-chloromethylpyrrolidines could be obtained as the major products for intramolecular chloroamination of N-substituted 4-penten-1-amines.18 The reaction could be carried out under mild conditions. However, it was difficult to get regiospecific products, and an additional step was generally required to fully convert the initially formed 2-chloromethylpyrrolidines to 3-chloropiperidines.19 In this communication, we wish to report our recent progress on transition metal-free intramolecular haloamination of 4-penten-1-amine and 5-hexen-1-amine substrates. High regioselectivity was realized, and the corresponding halogenated piperidines could be isolated in good yields.
In our course of searching for new reagents for intramolecular haloamination of 4-penten-1-amines, we found that intramolecular iodoamination products could be observed when zinc iodide was used. However, it was difficult to completely reproduce the results in follow-up studies. After a thorough work, we found that the reaction could take place when the reaction was carried out in untreated ethereal solvents such as crude THF or crude diethyl ether, but failed in sodium dried solvents. We reasoned that the reaction was induced by molecular iodine formed via the oxidation of zinc iodide by dissolved oxygen or possible peroxides formed during long time storage of the solvents.
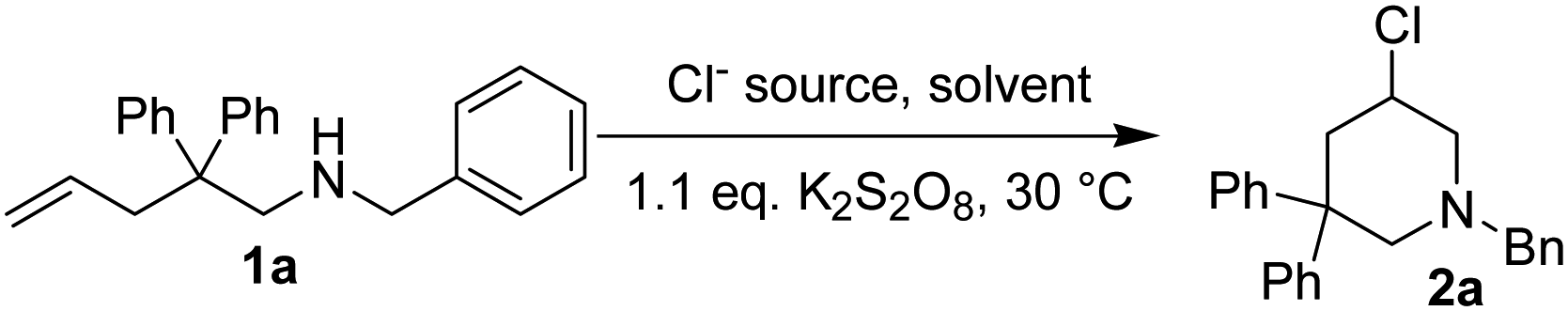
We then proposed a new route to 3-chloropiperidines as shown in Scheme 1. At first, iodine would react with the CC double bond of substrate 1a through normal electrophilic addition reaction, an iodonium 3-membered ring intermediate I was formed. Intramolecular nucleophilic attack of the nitrogen atom on the 3-membered ring of I produced the iodomethylpyrrolidine II as a temporary product. It was further converted to aziridinium intermediate III due to the good nucleophilicity of nitrogen atom and the good leaving property of I−. In the presence of excess amount of Cl−, nucleophilic ring opening of III by Cl− produced 3-chloropiperidine as the final products.12,13,16,17,20 Oxidation of I− with a suitable oxidant would regenerate iodine and complete the reaction cycle.
 | ||
| Scheme 1 Proposed reaction sequence for I2-mediated chloroamination. | ||
Several issues will have to be considered in order for chloroamination of substrate 1a to proceed successfully. The first issue is the iodine source. It could either be molecular iodine or other metal iodides. The second issue is the oxidant. The oxidant should meet the criteria such that it should be able to oxidize iodide to regenerate molecular iodine, but should not overoxidize iodide to OI− or IO2−. The third issue is a suitable chlorine source. It should be chosen such that it could gradually release Cl− to ensure a reasonable chloroamination, but should not participate any other reactions. The final issue is the reaction medium. It should have a reasonable solubility for both the oxidant and the chlorine source, no need to mention its ease of removal or handling.
Several easily available oxidants such as hydrogen peroxide, tert-butyl hydrogen peroxide (TBHP) and potassium persulfate were first tested. Hydrogen peroxide or TBHP failed to give good results possibly due to their overstrong oxidizing ability, and potassium persulfate was proven to be an ideal oxidant for the reaction. This was also in good agreement with the general knowledge that I− could be oxidized to molecular iodine in the presence of potassium persulfate.21 Potassium or zinc iodide gave similar results, and molecular iodine was proved to be more suitable for the reaction due to its good solubility in organic solvents.
After the screening of a suitable oxidant, other conditions such as iodine source, chlorine source and solvents were tested to optimize the reaction conditions, and the results are summarized in Table 1.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Chlorine source | Solvent | Time (h) | Isolated yield (%) |
| a In argon atmosphere. | |||||
| 1 | I2 (5) | LiCl (3 eq.) | THF | 7 | 84 |
| 2 | NaI (10) | LiCl (3 eq.) | THF | 48 | 78 |
| 3 | ZnI2 (10) | LiCl (3 eq.) | THF | 48 | 76 |
| 4 | I2 (2) | LiCl (3 eq.) | THF | 12 | 83 |
| 5 | I2 (1) | LiCl (3 eq.) | THF | 48 | 56 |
| 6 | I2 (2) | LiCl (3 eq.) | THF | 12 | 80a |
| 7 | I2 (2) | CaCl2 (3 eq.) | THF | 24 | 80 |
| 8 | I2 (2) | LiCl (3 eq.) | THF | 12 | 83 |
| 9 | I2 (2) | LiCl (3 eq.) | DCM | 12 | 18 |
| 10 | I2 (2) | LiCl (3 eq.) | EA | 12 | 51 |
| 11 | I2 (2) | LiCl (3 eq.) | CH3CN | 12 | 26 |
| 12 | I2 (2) | LiCl (3 eq.) | Toluene | 12 | 5 |
| 13 | I2 (2) | LiCl (3 eq.) | EtOH | 12 | 6 |
| 14 | I2 (2) | LiCl (3 eq.) | DMF | 12 | 22 |
| 15 | I2 (2) | LiCl (3 eq.) | DMSO | 12 | 1 |
| 16 | I2 (2) | LiCl (3 eq.) | Acetone | 12 | 20 |
| 17 | I2 (2) | LiCl (3 eq.) | 1,4-Dioxane | 12 | 19 |
As indicated in Table 1, THF was more superior to other solvents, and lithium chloride gave better results than calcium chloride did. The former was chosen as the chlorine source due to its slightly better solubility in THF. Other reason for using inorganic salts as chlorine source was their ease of removal. The amount of iodine was reduced to 2 mol% to avoid the possible formation of I3−. Reducing the amount of iodine will also suppress the possible formation of 3-iodopiperidine.
After establishing a general procedure for intramolecular chloroamination of 1a, other 4-penten-1-amines and 5-hexen-1-amines were tested to study the scope of the substrates, and the results are presented in Table 2.
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Reaction time (h) | Isolated yield (%) | ||
| R1 | R2 | ||||
| a NR. = no reaction. | |||||
| 1 | 1a | Ph | Bn | 12 | 83 |
| 2 | 1b | Ph | p-MeBn | 12 | 78 |
| 3 | 1c | Ph | p-MeOBn | 12 | 81 |
| 4 | 1d | Ph | p-FBn | 18 | 80 |
| 5 | 1e | Ph | p-O2NBn | 18 | 52 |
| 6 | 1f | Ph | iBu | 18 | 61 |
| 7 | 1g | Ph | nBu | 18 | 69 |
| 8 | 1h | Me | Bn | 24 | 30 |
| 9 | 1i | Me | nBu | 24 | 62 |
| 10 | 1j | –(CH2)5– | Bn | 24 | 36 |
| 11 | 1k | Allyl | Bn | 24 | 71 |
| 12 | 1l | H | Bn | 24 | 26 |
| 13a | 1m | Ts | Bn | 24 | N. R. |
| 14a | 1n | Ac | Bn | 24 | N. R. |
| 15 | 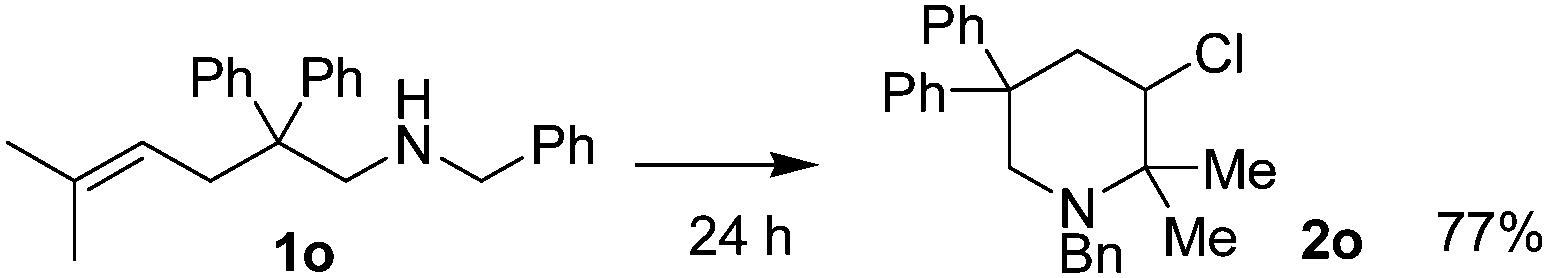 |
||||
| 16 |  |
||||
| 17 | 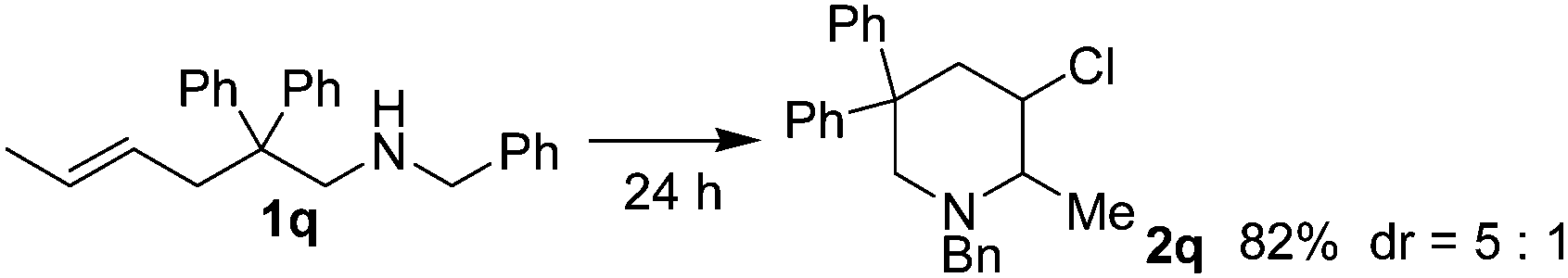 |
||||
 |
|||||
| 18 | 1r | Ph | Bn | 48 | 71 |
| 19 | 1s | Ph | p-MeBn | 48 | 66 |
| 20 | 1t | Ph | p-MeOBn | 48 | 69 |
| 21 | 1u | Ph | p-FBn | 48 | 62 |
| 22 | 1v | Ph | p-ClBn | 48 | 74 |
| 23 | 1w | –(CH2)5– | Bn | 48 | 65 |
| 24 | 1x | CH3 | Bn | 48 | 57 |

As shown in Table 2, Thorpe–Ingold effect was observed for the reaction. Substrates with diphenyl groups on the main chain (entries 1 to 7) gave good isolated yields, and substrates bearing small substituents (entries 8 to 11) or lacking of substituents on the main chain (entry 12) showed poor reactivity. In the case of substrate 1l, most of the starting material could be recovered after reacting for 24 hours. Sulfonamide or acetamide substrates (entries 13 and 14) failed to react due to low nucleophilicity of the nitrogen atom. Substrates with substituents on CC double bonds (entries 15 to 17) could also be converted, and acceptable diastereoselectivity was observed. 5-Hexen-1-amine substrates (entries 18 to 24) generally showed poor reactivity possibly due to the unfavorable entropy feature of the reaction. Increasing the amount of iodine, elevating the reaction temperature and elongating the reaction time led to the increase of isolated yields. Again, the course of the reaction was affected by the substituents on the main chain. Further, this type of substrates produced 2-chloromethylpiperidines instead of the 3-chloro-1-azacycloheptanes due to the stability of the six-member ring compounds.
In addition of the application of vicinal chloroamines in medicinal chemistry and organic chemistry, cyclic vicinal bromoamines were also found widespread applications in organic synthesis of a variety of N-heterocyclic compounds.22 To this end, bromoamination of substrate 1a was also tested to extend the application scope of the reaction. Conditions for chloroamination were first adopted, and results for screening of bromine source and reaction medium were summarized in Table 3.
|
|||
|---|---|---|---|
| Entry | Bromide | Solvent | NMR yielda |
| a Determined by crude 1H NMR.b Data in parentheses are isolated yield.c Reaction time = 48 hours. | |||
| 1 | LiBr | THF | <5 |
| 2 | MnBr2 | THF | 11 |
| 3 | ZnBr2 | THF | <5 |
| 4 | TBAB | THF | <5 |
| 5 | NBS | THF | 36 |
| 6 | Py·HBr | THF | 57 |
| 7 | Py·HBr | CH2Cl2 | 87 (73)b |
| 8 | Py·HBr | MeOH | <5 |
| 9 | Py·HBr | EtOAc | 30 |
| 10 | Py·HBr | CH2Cl2–EtOAc | 55 |
| 11c | Py·HBr | CH2Cl2 | >99 (88)b |

In contrast to chloroamination in which LiCl could be used as chlorine source, inorganic salts were not ideal bromine sources for the bromoamination reactions (Table 3, entries 1 to 3) possibly due the poor solubility of these metal bromides in reaction media. Conventional bromine sources such as TBAB or NBS (entries 4 and 5) also failed to give satisfactory results. Finally, pyridinium bromide was chosen as the bromine source. Good result was observed when the reaction was carried out in dichloromethane, possibly due to its good solubility in haloalkanes (entry 7). Complete conversion and excellent isolated yield was observed after 48 hours (entry 11).
After establishing a general procedure for bromoamination of substrate 1a, other substrates were also subjected to the same reaction to extend the application scope of the reaction, and the results were summarized in Table 4.As shown in Table 4, good to excellent results were obtained for substituted 4-penten-1-amine substrates (entries 1 to 8), and substrates lacking of efficient Thorpe–Ingold functional groups generally gave poor isolated yields at specified reaction time (entries 9 to 11). 5-Hexen-1-amine substrate gave low yield (entry 12) possibly due to the unfavourable entropy feather of the cyclization reaction.
|
|||
|---|---|---|---|
| Entry | Substrate | Isolated yield (%) | |
| R1 | R2 | ||
| 1 | Ph | Bn | 86 (3a) |
| 2 | Ph | p-MeBn | 90 (3b) |
| 3 | Ph | p-MeOBn | 86 (3c) |
| 4 | Ph | p-FBn | 83 (3d) |
| 5 | Ph | p-ClBn | 80 (3e) |
| 6 | Ph | p-O2NBn | 82 (3f) |
| 7 | Ph | iPr | 65 (3g) |
| 8 | Ph | iBu | 72 (3h) |
| 9 | Me | Bn | 63 (3i) |
| 10 | –(CH2)5– | Bn | 66 (3j) |
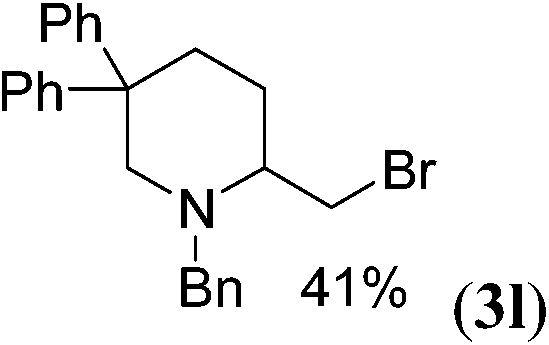
| 11 | H | Bn | 39 (3k) |
| 12 |  |
 |
|
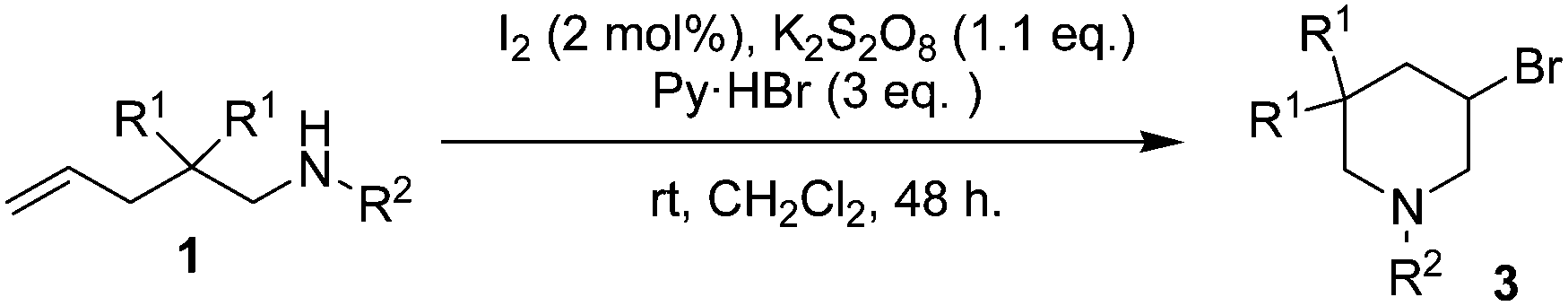
In summary, catalytic amount of molecular iodine can be used to promote intramolecular haloamination of unfunctionalized olefins. Good isolated yields were obtained for substituted 3-halopiperidines and 2-halomethylpiperidines. The reaction requires the presence of a suitable oxidant to regenerate the iodine. The current study provided an easy entry to a variety of substituted piperidines which could be used as important intermediates for both medicinal chemistry and organic synthesis.
Acknowledgements
We acknowledge the financial support from National Natural Science Foundation of China (NSFC 20972072, NSFC 21272121).Notes and references
- K. Pors, S. D. Shnyder, P. H. Teesdale-Spittle, J. A. Hartley, M. Zloh, M. Searcey and L. H. Patterson, J. Med. Chem., 2006, 49, 7013–7023 CrossRef CAS PubMed.
- (a) A. J. Blackman, C. Li, D. C. R. Hockless, B. W. Skelton and A. H. White, Tetrahedron, 1993, 49, 8645–8656 CrossRef CAS; (b) W. G. Kim, J. P. Kim, C. J. Kim, K. H. Lee and I. D. Yoo, J. Antibiot., 1996, 49, 20–25 CrossRef CAS; (c) L. Rahbæk and C. Christophersen, J. Nat. Prod., 1997, 60, 175–177 CrossRef.
- D. E. Horning and M. J. Muchowski, Can. J. Chem., 1974, 52, 1321–1330 CrossRef CAS.
- (a) R. Gottlich, Synthesis, 2000, 1561–1564 CrossRef CAS PubMed; (b) R. Gottlich and M. Noack, Tetrahedron Lett., 2001, 42, 7771–7774 CrossRef CAS; (c) G. Heuger, S. Kalsow and R. Gottlich, Eur. J. Org. Chem., 2002, 1848–1854 CrossRef CAS; (d) M. Noack and R. Gottlich, Eur. J. Org. Chem., 2002, 3171–3178 CrossRef CAS; (e) M. Noack and R. Gottlich, Chem. Commun., 2002, 536–537 RSC.
- Å. Sjöholm, M. Hemmerling, N. Pradeille and P. Somfai, J. Chem. Soc., Perkin Trans. 1, 2001, 891–899 RSC.
- M. R. Manzoni, T. P. Zabawa, D. Kasi and S. R. Chemler, Organometallics, 2004, 23, 5618–5621 CrossRef CAS.
- A. W. Lei, X. Y. Lu and G. S. Liu, Tetrahedron Lett., 2004, 45, 1785–1788 CrossRef CAS PubMed.
- F. E. Michael, P. A. Sibbald and B. M. Cochran, Org. Lett., 2008, 10, 793–796 CrossRef CAS PubMed.
- C. H. Hovelmann, J. Streuff, L. Brelot and K. Muniz, Chem. Commun., 2008, 2334–2336 RSC.
- G. Y. Yin, T. Wu and G. S. Liu, Chem.–Eur. J., 2012, 18, 451–455 CrossRef CAS PubMed.
- (a) H.-T. Huang, T. C. Lacy, B. Błachut, G. X. Ortiz and Q. Wang, Org. Lett., 2013, 15, 1818–1821 CrossRef CAS PubMed; (b) T. Wu, J. Cheng, P. Chen and G. Liu, Chem. Commun., 2013, 49, 8707–8709 RSC.
- N. De Kimpe and M. Boelens, J. Chem. Soc., Chem. Commun., 1993, 916–918 RSC.
- N. De Kimpe, M. Boelens, J. Piqueur and J. Baele, Tetrahedron Lett., 1994, 35, 1925–1928 CrossRef CAS.
- D. De Smaele and N. De Kimpe, J. Chem. Soc., Chem. Commun., 1995, 2029–2030 RSC.
- N. De Kimpe, M. Boelens and J. Contreras, Tetrahedron Lett., 1996, 37, 3171–3174 CrossRef CAS.
- H. A. Dondas and N. De Kimpe, Tetrahedron Lett., 2005, 46, 4179–4182 CrossRef CAS PubMed.
- E. R. Alonso, K. A. Tehrani, M. Boelens and N. De Kimpe, Synlett, 2005, 1726–1730 CAS.
- G. Q. Liu, W. Li and Y. M. Li, Adv. Synth. Catal., 2013, 355, 395–402 CAS.
- R.-L. Li, G.-Q. Liu, W. Li, Y.-M. Wang, L. Li, L. Duan and Y.-M. Li, Tetrahedron, 2013, 69, 5867–5873 CrossRef CAS PubMed.
- (a) R. H. Reitsema, J. Am. Chem. Soc., 1949, 71, 2041–2043 CrossRef CAS; (b) J. Cossy, C. Dumas and D. G. Pardo, Eur. J. Org. Chem., 1999, 1693–1699 CrossRef CAS.
- S. O. Rawling and J. W. Glassett, J. Phys. Chem., 1924, 29, 414–420 CrossRef.
- J. E. G. Kemp, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, pp. 469–513 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General procedure for transition metal-free haloamination reaction, characterization of new compounds, and copies of spectra data of the compounds. See DOI: 10.1039/c4ra00383g |
| This journal is © The Royal Society of Chemistry 2014 |