 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate and catalytic promiscuity of secondary metabolite enzymes: O-prenylation of hydroxyxanthones with different prenyl donors by a bisindolyl benzoquinone C- and N-prenyltransferase†
Sylwia Tarcza,
Xiulan Xieb and
Shu-Ming Li*a
aInstitut für Pharmazeutische Biologie und Biotechnologie, Philipps-Universität Marburg, Deutschhausstrasse 17A, 35037 Marburg, Germany. E-mail: shuming.li@staff.uni-marburg.de
bFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse, 35032 Marburg, Germany
First published on 7th April 2014
Abstract
Prenylated xanthones are secondary metabolites with a broad spectrum of biological activities including antimicrobial and antitumor activities. One xanthone O-prenyltransferase XptB has been identified in Aspergillus nidulans. Recently, we characterised a bisindolyl benzoquinone C- and N-prenyltransferase AstPT from Aspergillus terreus with unusually high substrate specificity towards both the prenyl donor dimethylallyl diphosphate and acceptor bisindolyl benzoquinone. In this study, we demonstrate the acceptance of a number of hydroxyxanthones by AstPT in the presence of not only dimethylallyl but also geranyl and farnesyl diphosphate. Structural elucidation by HR-MS and NMR analyses proved the O-prenylation of all thirteen isolated enzyme products with different prenyl moieties. These results demonstrated the remarkable substrate and catalytic promiscuity of AstPT, which was recognised as a specific enzyme prior to this study.
Introduction
Xanthones are naturally occurring secondary metabolites and widely distributed in plants, lichens, fungi and bacteria.1 The dibenzo-γ-pyrone core is supposed to be essential for their biological and pharmacological activities, which can be further modified by numerous substituents.2 For example, xanthones were reported to show cholinesterase inhibition activity and are therefore potential candidates for treatment of Alzheimer's disease.3 Antibacterial, antifungal and antiviral activities have also been reported for xanthones.4 Xanthones from plants and fungi are derived from different biosynthetic origins. Fungal xanthones are polyketides derived from acetate units while xanthones from plants are of mixed origin of both polyketide and shikimate pathways.1 However, prenyl moieties have to be attached by prenyltransferases in both sources. Decoration of xanthones by prenyl moieties often enhances the biological activities such as antioxidant, antibacterial, antifungal and antimalarial, antitumor and anti-HIV activities.5 Both dimethylallyl and geranyl moieties as well as structure features derived thereof have been found in xanthones from fungi.1 The prenyl moieties are attached to either a carbon atom of the aromatic core or an oxygen atom of a hydroxyl group. In ascomycetous fungi, xanthones have been found in different genera such as Aspergillus, Penicillium and Chaetomium.1 Despite the large number of prenylated xanthones only two xanthone prenyltransferases have been identified by gene deletion experiments in Aspergillus nidulans.6 XptB, as one of them, was successfully characterised biochemically and found to be responsible for the prenylation of 1,7-dihydroxy-6-methyl-8-hydroxymethylxanthone.7By using recombinant protein from E. coli, we demonstrated that AstPT from Aspergillus terreus was responsible for the transfer of a dimethylallyl moiety from dimethylallyl diphosphate (DMAPP) to N-1 and C-2 of asterriquinone D (AQ D, Fig. 1), a methylated bisindolyl benzoquinone derivative.8 In the presence of AQ D, AstPT accepted only DMAPP but not geranyl (GPP) or farnesyl diphosphate (FPP) as prenyl donor. This observation is consistent with the relatively high substrate specificity of the members of the DMATS superfamily9 to which AstPT also belongs. In contrast to most of the enzymes of the DMATS superfamily with relaxed substrate specificities towards aromatic substrates, AstPT did not accept tryptophan, tryptophan-containing cyclic dipeptides, hydroxynaphthalenes or flavonoids as prenylation substrates.8 Interestingly, product formation was detected in the incubation mixtures of AstPT with several hydroxyxanthones in the presence of DMAPP, GPP and FPP as prenyl donors. In this article we report its behavior towards hydroxyxanthones and the identification of the enzyme products as O-prenylated derivatives.
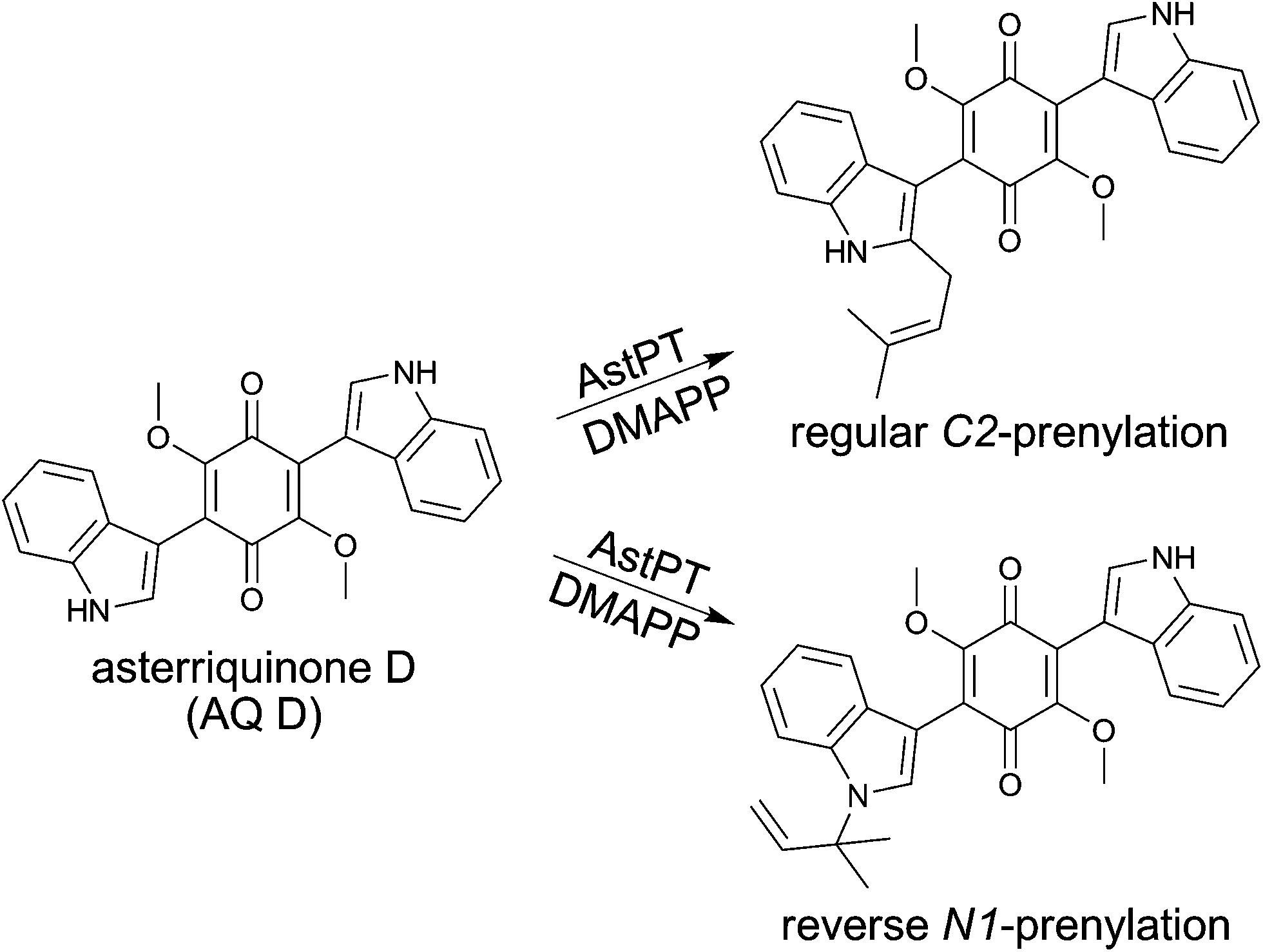 | ||
| Fig. 1 Regular and reverse prenylation of asterriquinone D by AstPT. | ||
Results and discussion
Acceptance of hydroxyxanthones by AstPT in the presence of different prenyl donors
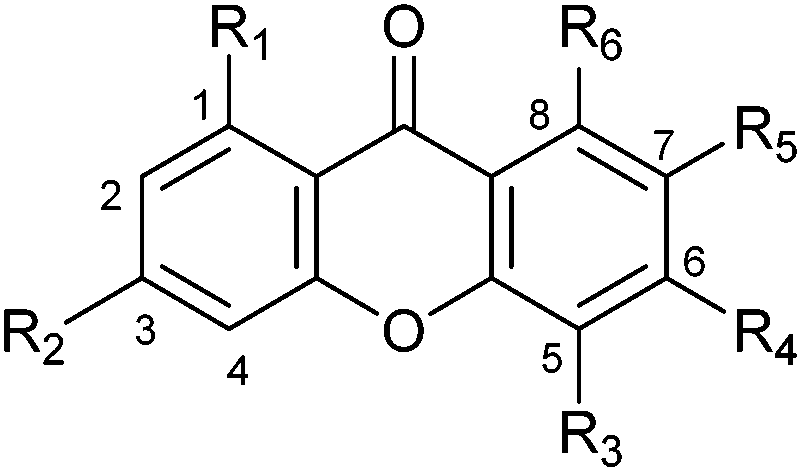
In comparison to the increased number of tryptophan and cyclic dipeptide prenyltransferases9 only one xanthone prenyltransferase XptB has been characterised biochemically.7 XptB is involved in the biosynthesis of (epi)shamixanthone in Aspergillus nidulans and catalysed O-prenylations of 1,7-dihydroxy-6-methyl-8-hydroxymethylxanthone and analogues.6,7 Our previous results showed that tryptophan and cyclic dipeptide prenyltransferases of the DMATS superfamily also used hydroxynaphthalenes and flavonoids as prenylation substrates.10,11 Hence, we hope to find enzymes for prenylation of xanthones.AstPT from Aspergillus terreus was shown to be responsible for the prenylation of AQ D.8 Although AstPT did not accept tryptophan or tryptophan-containing cyclic dipeptides as prenylation substrates, we carried out incubations of this enzyme with xanthone and ten hydroxylated derivatives (Table 1) in the presence of DMAPP, GPP or FPP. After incubation with 20 μg of AstPT in 100 μL assays at 37 °C for 16 h, the ethyl acetate extracts of the reaction mixtures were analysed on HPLC. HPLC analyses revealed notable product formation in the incubation mixtures of four hydroxyxanthones (1a–4a), not only with DMAPP but also with GPP and even with FPP (Fig. 2). In comparison to the results of negative controls with heat-inactivated protein, one additional predominant peak each was observed in the HPLC chromatograms of enzyme assays of 1a–3a with all tested prenyl donors and of 4a with GPP and FPP. In the assay of 4a with DMAPP three additional peaks were observed. No product formation was detected in the incubation mixtures with heat-inactivated protein (Fig. 2) or in the absence of the prenyl donors (data not shown). 1a was accepted as the best xanthone derivative with conversion yields of 23.1, 26.1 and 18.8% for DMAPP, GPP and FPP, respectively. 2a and 4a were accepted by AstPT with relative activities of 30 to 75% of those with 1a and the respective prenyl donors (Table 1). Remarkably, the relative activities in the assays with different prenyl donors for a given xanthone derivative did not change significantly. For 1a and 2a, GPP was slightly better accepted than DMAPP and FPP. In the case of 4a FPP was the best tested prenyl donor.
| Substrate |
|
Absolute conversion [%] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | R6 | DMAPP | GPP | FPP | |
| a Incubations were carried out using 20 μg of recombinant protein for 16 h at 37 °C. Conversion yields for substrates 1a–4a are calculated from peak areas of the substrate and products in HPLC chromatograms and their integrals in the 1H NMR spectra (as incubation mixtures for isolation of the enzyme products). n.d.: not determined. | |||||||||
| Xanthone | H | H | H | H | H | H | <0.1 | n.d. | n.d. |
| 3-Hydroxyxanthone | H | OH | H | H | H | H | <0.1 | n.d. | n.d. |
| 1-Hydroxy-6,8-dimethylxanthone | OH | H | H | CH3 | H | CH3 | 0.3 | n.d. | n.d. |
| 1,3-Dihydroxyxanthone | OH | OH | H | H | H | H | 0.9 | n.d. | n.d. |
| 1,7-Dihydroxy-6-methylxanthone (1a) | OH | H | H | CH3 | OH | H | 23.1 | 26.1 | 18.8 |
| 1,7-Dihydroxy-6,8-dimethylxanthone | OH | H | H | CH3 | OH | CH3 | <0.1 | n.d. | n.d. |
| 1,7-Dihydroxy-5,6,8-trimethylxanthone | OH | H | CH3 | CH3 | OH | CH3 | 0.4 | n.d. | n.d. |
| 1,7-Dihydroxy-6-methyl-8-hydroxy-methylxanthone | OH | H | H | CH3 | OH | CH2OH | <0.1 | n.d. | n.d. |
| 1,3,6-Trihydroxyxanthone (2a) | OH | OH | H | OH | H | H | 11.0 | 11.2 | 6.2 |
| 1,3,7-Trihydroxyxanthone (3a) | OH | OH | H | H | OH | H | 6.1 | 1.7 | 2.7 |
| 1,3,6,8-Tetrahydroxyxanthone (4a) | OH | OH | H | OH | H | OH | 11.8 | 8.4 | 14.0 |
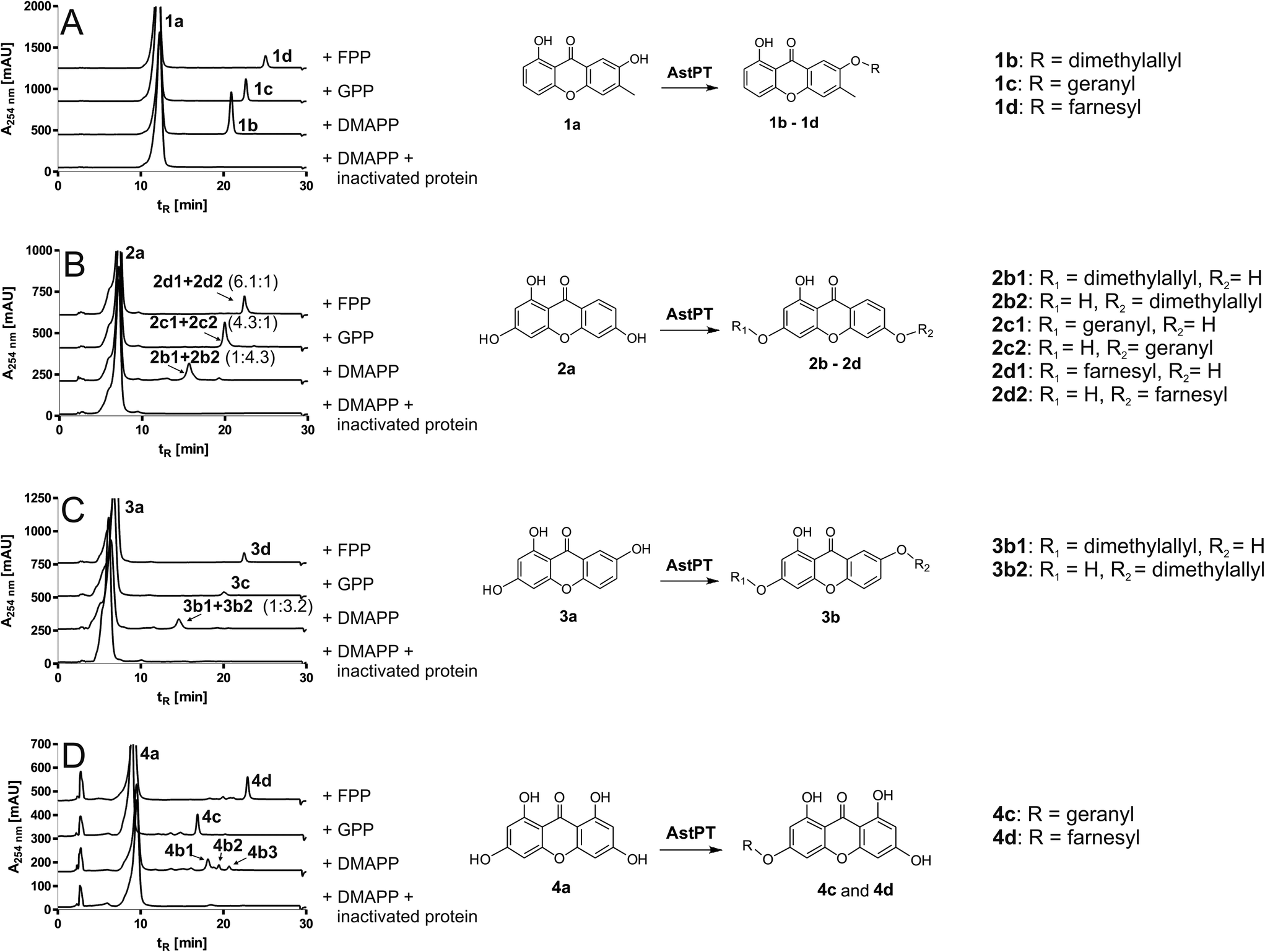 | ||
| Fig. 2 HPLC chromatograms of incubations of AstPT with hydroxyxanthones (left) and enzyme reactions catalysed by AstPT (right). Chromatograms are shown for incubations with 1,7-dihydroxy-6-methylxanthone (1a, (A)), 1,3,6-trihydroxyxanthone (2a, (B)), 1,3,7-trihydroxyxanthone (3a, (C)) and 1,3,6,8-tetrahydroxyxanthone (4a, (D)), using inactivated protein or DMAPP, GPP or FPP as prenyl donor. Incubations were carried out at 37 °C for 16 h and subsequently extracted three times with ethyl acetate. | ||
The intriguing results of AstPT with hydroxyxanthones prompted us to compare its amino acid sequence with that of XptB again and to carry out enzyme reactions of XptB with its natural substrate 1,7-dihydroxy-6-methyl-8-hydroxymethylxanthone6,7 and the natural substrate of AstPT AQ D in the presence of DMAPP, GPP and FPP. AstPT and XptB share a sequence identity of only 23% on the amino acid level (Table S1†). XptB did not accept AQ D as aromatic substrate by using DMAPP, GPP or FPP as prenyl donor at 37 °C for 16 h. Product formation was only detected in the XptB assay of 1,7-dihydroxy-6-methyl-8-hydroxymethylxanthone with DMAPP, but not with GPP or FPP (Fig. S1†).
Structural elucidation of enzyme products
For structure elucidation, the product peaks were isolated from incubation mixtures of 1a–4a with DMAPP, GPP and FPP on HPLC and subjected to MS and NMR analyses. Nine peaks, 1b–1d, 2b–2d, 3b, 4c and 4d were isolated in substantial amounts so that interpretable HR-MS and NMR spectra were obtained (Tables 2 and S2–S5†) for structure elucidation. Molecular ions in positive HR-EI-MS mode confirmed the monoprenylation in the isolated products, as the masses of the products with DMAPP were 68 Da, with GPP 136 Da and with FPP 204 Da larger than those of the respective substrates. Inspection of the 1H NMR spectra of the enzyme products revealed that all signals for the aromatic protons were still present as well as the corresponding signal of the methyl group in 1a indicating that the prenylations have taken place at one of the hydroxyl groups. This also coincided with the appearance of the signals for H-1′ of the prenyl moieties at 4.69 to 4.79 ppm with coupling constants between 6–7 Hz, which is typical for an O-prenylation.12 The signals of the H-2′ appeared as triplets from 5.35 to 5.54 ppm. The signals of the methyl groups of the prenyl moieties (two for dimethylallyl, three for geranyl and four for farnesyl moiety) were detected between 1.55 and 1.86 ppm. The signals for additional olefinic protons (one for geranyl and two for farnesyl moiety) appeared as triplets between 5.03 and 5.54 ppm. These features proved that they were just attached to hydroxyl groups and not further modified, e.g. by cyclisation.| Compound | Chemical formula | [M]+ | Deviation [ppm] | |
|---|---|---|---|---|
| Calculated | Measured | |||
| 1b | C19H18O4 | 310.1205 | 310.1227 | 7.1 |
| 1c | C24H26O4 | 378.1831 | 378.1817 | −3.7 |
| 1d | C29H34O4 | 446.2487 | 446.2457 | −6.7 |
| 2b1 + 2b2 | C18H16O5 | 312.0998 | 312.0994 | −1.3 |
| 2c1 + 2c2 | C23H24O5 | 380.1624 | 380.1632 | 2.1 |
| 2d1 + 2d2 | C28H32O5 | 448.2224 | 448.2250 | 5.8 |
| 3b1 + 3b2 | C18H16O5 | 312.0998 | 312.0979 | −6.1 |
| 4c | C23H24O6 | 396.1573 | 396.1595 | 5.6 |
| 4d | C28H32O6 | 464.2199 | 464.2158 | −8.8 |
Comparison of the NMR data of 1b, 1c and 1d revealed that the aromatic protons have identical chemical shifts with nearly same coupling constants indicating the same prenylation position in their structures. Relatively strong downfield shift of +0.06 ppm was observed for the signals of H-5, probably caused by a prenylation at OH-7 rather than OH-1. To confirm the prenylation position, HMBC spectrum was taken for 1d. Correlations were clearly observed between H-1′ and C-7, proving the attachment of the farnesyl moiety to OH-7 (Fig. S15 and S16†). Therefore, 1d was identified as 6-methyl-7-farnesyloxy-1-hydroxyxanthone (Fig. 2). The position of the prenyl moieties in 1b and 1c was assigned analogously as the same changes were found in these 1H NMR spectra as for 1d. Hence, 1b and 1c were identified as 6-methyl-7-dimethylallyloxy-1-hydroxyxanthone and 6-methyl-7-geranyl-oxy-1-hydroxyxanthone, respectively (Fig. 2).
Inspection of the 1H NMR spectra of the isolated peaks 2b–2d revealed the presence of two prenylated products each (Table S3, Fig. S7–S9†). The ratios of 2b1 to 2b2 from the incubation mixture of 2a with DMAPP and 2c1 to 2c2 from that of 2a with GPP were found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.3 and 4.3:1, respectively. Similar ratio of 6.1:1 was calculated for 2d1 to 2d2 from the incubation with FPP. Unfortunately, these compounds could not be separated from each other and their structures were elucidated by interpretation of the NMR spectra of the mixtures. This was possible due to the different intensities of the signals for protons of both compounds. The chemical shifts of H-7 and H-8 in 2b1, 2c1 and 2d1 have almost not changed in comparison to those of 2a indicating that the prenylation was unlikely at OH-6. This conclusion was supported by the significant changes of chemical shifts for H-2, H-4 and H-5. The prenylation position in 2b1, 2c1 and 2d1 was therefore assigned to OH-3 (Fig. 2). In comparison to those of 2b1, 2c1 and 2d1, the chemical shifts for H-5 and H-7 in 2b2, 2c2 and 2d2 differed clearly from those in 2a. In contrast, only slight changes have been observed for H-2. Therefore, the prenyl moieties in 2b2, 2c2 and 2d2 were assigned to OH-6.
:4.3 and 4.3:1, respectively. Similar ratio of 6.1:1 was calculated for 2d1 to 2d2 from the incubation with FPP. Unfortunately, these compounds could not be separated from each other and their structures were elucidated by interpretation of the NMR spectra of the mixtures. This was possible due to the different intensities of the signals for protons of both compounds. The chemical shifts of H-7 and H-8 in 2b1, 2c1 and 2d1 have almost not changed in comparison to those of 2a indicating that the prenylation was unlikely at OH-6. This conclusion was supported by the significant changes of chemical shifts for H-2, H-4 and H-5. The prenylation position in 2b1, 2c1 and 2d1 was therefore assigned to OH-3 (Fig. 2). In comparison to those of 2b1, 2c1 and 2d1, the chemical shifts for H-5 and H-7 in 2b2, 2c2 and 2d2 differed clearly from those in 2a. In contrast, only slight changes have been observed for H-2. Therefore, the prenyl moieties in 2b2, 2c2 and 2d2 were assigned to OH-6.
Using 3a as substrate, interpretable spectra were only obtained for the products of the incubation mixture with DMAPP (Table S4, Fig. S11†). This sample was found to be a mixture of 3b1 and 3b2 with a ratio of 1:3.2. Comparison of the spectrum of 3b1 with that of 3a, significant changes of chemical shifts were found for H-2 and H-4 and nearly no changes for H-5, H-6 and H-8. In the spectrum of 3b2 obvious changes were found for H-5 and H-6 with upfield shifts of 0.05 and 0.06 ppm to that of 3a, respectively. These proved the prenylation at OH-3 in 3b1 and at OH-7 in 3b2.
4a has a symmetric structure; the 1H NMR spectrum shows therefore only two coupling doublets of two aromatic protons (Table S5, Fig. S12†). Interpretable spectra were obtained for 4c from incubation mixture with GPP and 4d with FPP. Two different coupling systems were observed in the spectra of both compounds (Fig. S13 and S14†). In the spectra of 4c and 4d, obvious changes of chemical shifts were found for H-2 and H-4. Thus, the prenylation position was assigned to OH-3 in both cases.
Determination of kinetic parameters of AstPT
In order to determine the affinity of AstPT towards 1a–4a, we incubated these compounds with increased concentrations (Fig. S17–S20†). Kinetic parameters for the reaction with 2a could not be determined because inhibition occurred before the maximal velocity was reached under our assay conditions. The reactions of AstPT with 1a, 3a and 4a in the presence of DMAPP apparently followed Michaelis–Menten kinetics. Kinetic parameters including Michaelis–Menten constant (KM) and turnover number (kcat) were calculated from Eadie–Hofstee, Hanes–Woolf and Lineweaver–Burk plots and are listed in Table 3. 1a with a KM at 17.3 μM was found to be the best accepted hydroxyxanthone. For 3a and 4a, KM values were determined at 168 and 28.6 μM, respectively. These values are much lower than the KM of 463 μM for AstPT with AQ D8 The reaction velocity of AstPT with AQ D was however at least 160-fold as those of hydroxylxanthones, e.g. kcat of 0.16 s−1 for AQ D versus 0.001 s−1 for 1a. Using 1a as aromatic substrate, KM values were determined for DMAPP and GPP at 97.3 and 12.1 μM, respectively (Fig. S21 and S22†).| Compound | KM [μM] | kcat [s−1] | kcat/KM [s−1 M−1] |
|---|---|---|---|
| a Kinetic parameters for aromatic substrates were determined using DMAPP as prenyl donor.b Kinetic parameters for prenyl donors were determined using 1a as aromatic substrate.c Kinetic parameters were not determined, because maximal velocity could not be reached before inhibition occurred under our conditions. | |||
| 1aa | 17.3 | 0.001 | 55.2 |
| 2aa | —c | —c | —c |
| 3aa | 168 | 0.0007 | 4.2 |
| 4aa | 28.6 | 0.0003 | 8.6 |
| DMAPPb | 97.3 | 0.001 | 8.5 |
| GPPb | 12.1 | 0.002 | 169.6 |
| FPPb | —c | —c | —c |
Comparison of active sites in prenyltransferases of the DMATS superfamily
Prenyltransferases of the DMATS superfamily often show low sequence identities of approximate 30% to each other (Table S1†), but accept similar aromatic compounds as substrates.9 In order to identify conserved amino acid residues, a multiple sequence alignment with AstPT and seven known prenyltransferases including XptB was performed (Fig. S24†) and indicated that only a few amino acids are conserved in the examined sequences. This is in good agreement with our previous observations. Usually, prenylation position (C-, N- or O-atom) or pattern (reverse or regular) could not be predicted by sequence analysis and comparison.13 Analysis of the crystal structures of FgaPT2,14 FtmPT1,15 CdpNPT16 and AnaPT17 revealed that these enzymes share a similar secondary core structure with ten antiparallel β-strands, which are surrounded by a ring of α-helices. Comparison with the available structures revealed that tyrosine residues involved in the stabilisation of the diphosphate moiety of DMAPP were highly conserved. These residues can also be found in AstPT, i.e. Y252, Y338, Y402 and Y406. Furthermore, mutagenesis experiments have shown that two conserved lysine residues, corresponding to K180 and K250 in AstPT, play an essential role in the binding of the prenyl donor.18 Two conserved arginine residues were also shown to be involved in the binding of diphosphate (Fig. S24†). The importance of R113 in the structure of FtmPT1, corresponding to R95 in AstPT, was demonstrated by Jost et al.15 The second conserved arginine residue was shown to be essential for the catalytic activity in FgaPT2 (R257), but not in FtmPT1 (R292).18 This residue is also present in AstPT at position R248. Stabilisation of the nitrogen atom of the indole ring during prenylation was performed by a glutamate residue in FgaPT2 (E89),14 FtmPT1 (E102),15 CdpNPT (E116)16 and AnaPT (E111).17 TdiB and AstPT also catalyse prenylations at the indole ring.8,19 In TdiB the glutamate residue is replaced by aspartate (D67), which can undergo similar interactions. This residue in AstPT is an asparagine (N81).Discussion
Most fungal prenyltransferases of the DMATS superfamily are known to have relaxed substrate specificities and accepted a series of aromatic compounds as prenyl acceptors. These include not only structures which are closely related to their natural substrates such as tryptophan or tryptophan-containing cyclic dipeptides, but also significantly different substances like hydroxynaphthalenes or flavonoids.9 On the other hand, they utilise usually only DMAPP as a prenyl donor. Until now only one known enzyme from the DMATS superfamily, i.e. VrtC from Penicillium aethiopicum, used GPP but not DMAPP as prenyl donor.20 Recently, we demonstrated that AnaPT from Neosartorya fischeri accepted both DMAPP and GPP as prenyl donors and catalysed prenylations of tryptophan-containing cyclic dipeptides with different pattern and positions.Very recently, we successfully cloned, overexpressed and purified AstPT from Aspergillus terreus and showed that this enzyme catalysed the transfer of a dimethylallyl moiety from DMAPP to AQ D, a methylated bisindolyl benzoquinone.8 AstPT was proven to be very specific for its prenyl donor DMAPP and acceptor AQ D. It accepted no further aromatic substrates of known prenyltransferases and no other prenyl donors such as GPP and FPP. In this study we demonstrated that AstPT was in fact able to transfer prenyl moieties not only to AQ D, but also to hydroxyxanthones. Both AQ D (Fig. 1) and hydroxyxanthones share similar structural features, i.e. a planar aromatic system as well as carbonyl groups. As hydroxyl groups in AQ D are blocked by methyl groups, a prenylation of these groups is not possible. On the other hand, hydroxyxanthones contain no heterocyclic nitrogen and an N-prenylation is not possible. Recently, the O-prenyltransferase SirD was shown capable of transferring prenyl moieties to N-, C- or S-atoms, proving that the prenylation position can be switched in the presence of unnatural substrates.21 A multiple sequence alignment of AstPT and seven prenyltransferases of the DMATS superfamily revealed that several conserved prenyl diphosphate binding moieties are present in the sequence of AstPT, but no suggestions can be made for the binding of the aromatic substrates or a mechanism for substrate activation. The results obtained in this study raised an important question, if bisindolyl benzoquinone and xanthone prenyltransferases share close biochemical relationships. Therefore, we tested the acceptance of AQ D by the xanthone O-prenyltransferease XptB from Aspergillus nidulans in the presence of different prenyl donors. No activity was observed in these assays (Fig. S1†).
Another interesting feature of AstPT refers to the acceptance of DMAPP, GPP and FPP in the presence of hydroxyxanthones. GPP was an even better substrate than DMAPP (Fig. 2, Tables 1 and 3). As aforementioned, GPP was not accepted by AstPT in the presence of AQ D. For the reaction of AstPT with AQ D and DMAPP, KM and kcat were determined at 33.5 μM and 0.02 s−1, respectively.8 In comparison, these values are 12.1 μM and 0.002 s−1 for the reaction with 1a and GPP providing evidence that AQ D is very likely the natural substrate for AstPT rather than hydroxyxanthones.
From the structures of the enzyme products (Fig. 2), it is obvious that OH-1 of xanthones was a poor prenylation position for AstPT, probably due to a steric hindrance by the keto group. No O1-prenylated derivative was identified in this study. This hypothesis explains the fact that only one main product was found in the enzyme assays of 1a with DMAPP, GPP and FPP each as well as those of 4a with GPP and FPP. In contrast, two products each with prenylations at OH-3, OH-6 or OH-7 were identified in the reaction mixtures of 2a and 3a. With DMAPP as prenyl donor, O6- or O7-prenylated derivative was predominant product in the reaction mixtures of 2a and 3a. In the reaction mixtures of 2a with GPP and FPP, O3-prenylated derivatives are predominant products.
Experimental
Chemicals
DMAPP, GPP and FPP were prepared according to the method described for GPP by Woodside.22 Hydroxyxanthones were synthesised as described before.7Computer-assisted sequence analysis
Alignments of amino acid sequences were carried out by using the program BLAST (http://blast.ncbi.nlm.nih.gov). Multiple sequence alignments of prenyltransferases were performed using Clustal Omega (1.2.1) (https://www.ebi.ac.uk/Tools/msa/clustalo/).Bacterial strains, plasmids and culture conditions
E. coli XL1-Blue MRF' (Stratagene, Amsterdam, the Netherlands) were used for gene expression. Bacteria harbouring pST248 or pDP0087 were cultivated at 37 °C in liquid or on solid lysogeny broth (LB) with agar (1.5% w/v) supplemented with carbenicillin (50 μg mL−1) for selection of recombinant E. coli strains.23
Overproduction, purification and analysis of recombinant protein
Overproduction and purification of recombinant AstPT-His6 or XptB-His6 from cultures of E. coli XL1-Blue MRF' cells harbouring pST24 or pDP008 were carried out as described previously.7,8 The purity of the recombinant proteins was analysed on SDS-PAGE according to the method of Laemmli24 with 12% polyacrylamide gels by using Coomassie Brilliant Blue G 250 for staining. Protein quantification was carried out according to the method of Bradford.25Enzyme assays and determination of kinetic parameters
The enzyme reaction mixtures (100 μL) contained 50 mM Tris–HCl (pH 7.5), 5 mM CaCl2, 0.5 mM xanthone derivative, 2 mM DMAPP, 0.5 mM GPP or FPP, 2.5% (v/v) DMSO, 2.0% (v/v) glycerol and 20 μg of purified recombinant AstPT-His6 (4.1 μM). The reaction mixtures were incubated at 37 °C for 16 h. The enzyme reaction mixtures for testing the acceptance of AQ D by XptB contained 50 mM Tris–HCl (pH 7.5), 5 mM MnCl2, 0.5 mM AQ D, 1 mM DMAPP, GPP or FPP, 2.5% (v/v) DMSO, 1.5% (v/v) glycerol and 20 μg of purified recombinant XptB-His6. For determination of the kinetic parameters for hydroxyxanthones, the assays contained DMAPP (2 mM) and hydroxyxanthones at final concentrations of up to 0.5 mM (1a) or 1.0 mM (2a, 3a, 4a). For determination of kinetic parameters of the prenyl donors, the assays contained 1a (0.5 mM) and prenyl donors at final concentrations of up to 3 mM. Incubations were carried out for 60 min (1a, DMAPP, GPP), 120 min (2a, 4a, FPP) or 150 min (2a) and contained 10 μg (1a, 3a, 4a, DMAPP, GPP) or 20 μg AstPT (2a, FPP). The enzyme reactions were stopped by triple extraction with 200 μL ethyl acetate. After evaporation of the combined organic phases to dryness, the residue was dissolved in 20 μL DMSO, diluted with 100 μL methanol (for hydroxyxanthones) or acetonitrile (for AQ D) and analysed by HPLC as described below. Two independent incubations were carried out routinely. Kinetic parameters were calculated as average values from Eadie–Hofstee, Hanes–Woolf and Lineweaver–Burk plots using GraphPad Prism 4.For isolation and structure elucidation of the enzyme products, incubations were carried out in 50 mL at 37 °C for 16 h. The incubation mixtures contained 50 mM Tris HCl (pH 7.5), 5 mM CaCl2, 0.5 mM hydroxyxanthone derivative, 2 mM DMAPP, 0.5 mM GPP or FPP, 2.5% (v/v) DMSO, 2.6% (v/v) glycerol and 12.0 mg of purified recombinant protein.
Conversion yields were calculated from the peak area of substrates and enzyme products in the HPLC chromatogram. Yields were corrected according to the ratio between substrates and products taken from the peak area from HPLC chromatograms and integrals from 1H NMR spectra of the incubation mixtures for isolation of enzyme products.
HPLC conditions for analysis and isolation of enzyme products
The enzyme products of the incubation mixtures were analysed by HPLC on an Agilent series 1200 system (Agilent Technologies, Santa Clara, CA, USA) by using a Multospher 120 RP 18-5 column (250 × 4 mm, 5 μm, CS-Chromatographie Service, Langerwehe, Germany) at a flow rate of 1 mL min−1. Water (solvent A) and methanol (solvent B) were used as solvents for analysis of incubation mixtures of hydroxyxanthones. A linear gradient of 65–100% (v/v) solvent B in 20 min was used. The column was then washed with 100% solvent B for 5 min and equilibrated with 65% (v/v) solvent B for 5 min. Detection was carried out by a photo diode array detector and absorptions at 254 nm were illustrated.For analysis of incubation mixtures of AQ D and XptB, water (solvent A) and acetonitrile (solvent B) were used as solvents. A linear gradient of 50–100% (v/v) solvent B in 20 min was used. The column was then washed with 100% solvent B for 5 min and equilibrated with 50% (v/v) solvent B for 5 min. Detection was carried out by a photo diode array detector and absorptions at 277 nm were illustrated.
For isolation of the enzyme products, the same HPLC equipment was used. Isolation of 1b and 2b1 + 2b2 was carried out on a Multospher 120 RP 18-5 column (250 × 4 mm, 5 μm, CS-Chromatographie Service) at a flow rate of 1 mL min−1. The same method was used as for the analysis of the enzyme assays. All other enzyme products were isolated on a Multospher 120 RP 18 HP column (250 × 10 mm, 5 μm, CS-Chromatographie Service). For isolation of 2c1 + 2c2 and 3b1 + 3b2, a linear gradient of 80–100% (v/v) solvent B in 15 min was followed by 100% (v/v) solvent B for 10 min at a flow rate of 2.5 mL min−1. The column was then equilibrated with 80% (v/v) solvent B for 5 min. For isolation of 2d1 + 2d2 and 4c, the elution profile was initiated with 90% (v/v) solvent B for 2 min at a flow rate of 3 mL min−1. Subsequently, a linear gradient of 90–100% (v/v) solvent B in 12 min was used and followed by 100% (v/v) solvent B for 5 min. The column was then equilibrated with 90% (v/v) solvent B for 5 min. The enzyme products 4d, 1c and 1d were isolated using 100% (v/v) solvent B in an isocratic run for 20 min (4d), 25 min (1c) or 33 min (1d) at a flow rate of 2.5 mL min−1.
Spectroscopic analysis of the enzyme products
The isolated enzyme products were dissolved in methanol and analysed by high-resolution electron impact mass spectrometry (HR-EI-MS) on an AutoSpec instrument (Micromass, Manchester, UK). Positive HR-EI-MS data of the enzyme products are listed in Table 2.For NMR analysis, the isolated products were dissolved in 0.75 mL of (CD3)2CO (Euriso-Top, Saarbrücken, Germany). Spectra were recorded at room temperature on a JEOL ECA-500 spectrometer (JEOL, Akishima, Tokyo, Japan). Heteronuclear multiple bond correlation (HMBC) spectrum of 1d in 0.30 mL (CD3)2SO (Euriso-Top) was recorded with standard methods26 on a Bruker Avance 500 MHz spectrometer. Spectra were processed with MestReNova.6.0.2 (Metrelab Research, Santiago de Compostella, Spain) and chemical shifts were referenced to the signal of (CD3)2CO at 2.05 ppm or to the 1H (2.50 ppm) and 13C signal (39.52 ppm) of (CD3)2SO. NMR data are given in Tables S2–S5.†
Conclusions
In this study, we demonstrated the substrate and catalytic promiscuity of the recently identified bisindolyl benzoquinone N- and C-prenyltransferase AstPT by identification of the O-prenylation of hydroxyxanthones in the presence of DMAPP, GPP and FPP. Our results reported here provided an example that the acceptance of unusual aromatic substrates has still to be discovered for the increasing group of the prenyltransferases of the DMATS superfamily. Furthermore, different prenyl donors can be used in the presence of different acceptors and therefore unnatural aromatic substrates should also be assayed with unnatural prenyl donors.Acknowledgements
We thank Dr Edyta Stec for synthesis of GPP and FPP, Lena Ludwig for synthesis of DMAPP and hydroxyxanthones, Nina Zitzer for taking MS. This work was supported by the Deutsche Forschungsgemeinschaft (Grant Li844/4-1 to S.-M. Li).References
- K.-S. Masters and S. Bräse, Chem. Rev., 2012, 112, 3717–3776 CrossRef CAS PubMed.
- M. M. Pinto, M. E. Sousa and M. S. Nascimento, Curr. Med. Chem., 2005, 12, 2517–2538 CrossRef CAS.
- J. Qin, W. Lan, Z. Liu, J. Huang, H. Tang and H. Wang, Chem. Cent. J., 2013, 7, 78 CrossRef PubMed.
- M. E. Rateb and R. Ebel, Nat. Prod. Rep., 2011, 28, 290–344 RSC.
- M. M. M. Pinto and R. A. P. Castanheiro, Curr. Org. Chem., 2009, 13, 1215–1240 CrossRef CAS.
- J. F. Sanchez, R. Entwistle, J. H. Hung, J. Yaegashi, S. Jain, Y. M. Chiang, C. C. Wang and B. R. Oakley, J. Am. Chem. Soc., 2011, 133, 4010–4017 CrossRef CAS PubMed.
- D. Pockrandt, L. Ludwig, A. Fan, G. M. Konig and S.-M. Li, ChemBioChem, 2012, 13, 2764–2771 CrossRef CAS PubMed.
- S. Tarcz, L. Ludwig and S.-M. Li, ChemBioChem, 2014, 15, 108–116 CrossRef CAS PubMed.
- X. Yu and S.-M. Li, Methods Enzymol., 2012, 516, 259–278 CAS.
- X. Yu, X. Xie and S.-M. Li, Appl. Microbiol. Biotechnol., 2011, 92, 737–748 CrossRef CAS PubMed.
- X. Yu and S.-M. Li, ChemBioChem, 2011, 12, 2280–2283 CrossRef CAS.
- H.-X. Zou, X. Xie, X.-D. Zheng and S.-M. Li, Appl. Microbiol. Biotechnol., 2011, 89, 1443–1451 CrossRef CAS PubMed.
- N. Steffan, A. Grundmann, W.-B. Yin, A. Kremer and S.-M. Li, Curr. Med. Chem., 2009, 16, 218–231 CrossRef CAS.
- U. Metzger, C. Schall, G. Zocher, I. Unsöld, E. Stec, S.-M. Li, L. Heide and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309–14314 CrossRef CAS PubMed.
- M. Jost, G. Zocher, S. Tarcz, M. Matuschek, X. Xie, S.-M. Li and T. Stehle, J. Am. Chem. Soc., 2010, 132, 17849–17858 CrossRef CAS PubMed.
- J. M. Schuller, G. Zocher, M. Liebhold, X. Xie, M. Stahl, S.-M. Li and T. Stehle, J. Mol. Biol., 2012, 422, 87–99 CrossRef CAS PubMed.
- X. Yu, G. Zocher, X. Xie, M. Liebhold, S. Schütz, T. Stehle and S.-M. Li, Chem. Biol., 2013, 20, 1492–1501 CrossRef CAS PubMed.
- E. Stec, N. Steffan, A. Kremer, H. Zou, X. Zheng and S.-M. Li, ChemBioChem, 2008, 9, 2055–2058 CrossRef CAS PubMed.
- C. J. Balibar, A. R. Howard-Jones and C. T. Walsh, Nat. Chem. Biol., 2007, 3, 584–592 CrossRef CAS PubMed.
- Y. H. Chooi, P. Wang, J. Fang, Y. Li, K. Wu, P. Wang and Y. Tang, J. Am. Chem. Soc., 2012, 134, 9428–9437 CrossRef CAS PubMed.
- J. D. Rudolf and C. D. Poulter, ACS Chem. Biol., 2013, 8, 2707–2714 CrossRef CAS PubMed.
- A. B. Woodside, Z. Huang and C. D. Poulter, Org. Synth., 1988, 66, 211–215 CrossRef CAS.
- J. Sambrook and D. W. Russell, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 2001 Search PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- S. Berger and S. Braun, 200 and More NMR Experiments. A Practical Course, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00337c |
| This journal is © The Royal Society of Chemistry 2014 |