Novel polyoxadiazoles with non-coplanar ortho-linked structures as highly CO2 permselective membranes
E. Abouzari-Lotf*a,
A. Shockravib,
A. Rafieimaneshb,
M. Saremib,
A. Javadib and
M. M. Nasef*a
aInstitute of Hydrogen Economy, Energy Research Alliance, International Campus, Universiti Teknologi Malaysia, 54100 Kuala Lumpur, Malaysia. E-mail: ebrahim@ic.utm.my; mahmoudeithar@cheme.utm.my
bFaculty of Chemistry, Kharazmi University, Mofatteh Ave. No.49, Postal Code 15614, Tehran, Iran
First published on 4th April 2014
Abstract
A novel class of polyoxadiazole membranes containing non-coplanar 1,1′-thiobis(2-naphthoxy) (S-BINOL) groups was synthesized using a versatile route and tested for CO2 separation. Dihydrazide monomer was obtained in a two-step high-yielding procedure and used in polycondensation reaction with terephthaloyl chloride to prepare a polyhydrazide, which was converted into amorphous and soluble polyoxadiazoles having an inherent viscosity of around 1 dL g−1 by a simple thermal cyclization without using any solvent or in solvent cyclization. The ortho-linked sulfide groups of the obtained polymers were subsequently oxidized to yield sulfone-containing polymers. Polyoxadiazole membranes demonstrated comparable CO2 separation properties to those of imide based polymers and almost better performance than commercial and modified polyethersulfones, and the previously reported polyoxadiazoles. The sulfone-containing polyoxadiazole membranes exhibited higher permeability, lower selectivity, and enhanced plasticization pressure compared to the corresponding sulfide containing ones.
Introduction
Membrane-mediated separations have been emerged as fast-growing separation technologies and have become vital components in various energy related applications.1,2 Polymeric membranes are the first and most common commercial membranes used for a variety of industrial gas separation processes including O2/N2 (e.g., nitrogen generation) and CO2/CH4 (e.g., natural gas enrichment). Membrane-based separation processes inherently provide low cost, high energy efficiency, design flexibility, and ease of operation. Among membrane gas separation processes, removal of CO2 from natural gas is a relatively new technology that is competing with various well-established conventional gas processing technologies such as absorption and adsorption. While there are huge demands and market potentials for membrane based CO2 separation, the persisting challenge remains in the development of durable membranes with enhanced selectivity and permeability.3,4 Therefore, research for advanced polymeric membranes with enhanced permselectivity and sufficient thermal, chemical, and mechanical stability at high temperature and pressure is ever receiving growing attention in the field of membrane science and technology.5,6As reported by Robeson,7,8 there is a general trade-off between the gas permeability and selectivity of polymeric materials. More selective membranes are often less permeable and vice versa. In this regard, several structure–property relationship studies have identified the best combinations of polymer structures resulting in high selectivity and permeability.7–10 Accordingly, two designing principles have been proposed for improving the selectivity and diffusivity of membranes:11–14 (i) Simultaneous increase in interchain separation (inhibition of intersegmental packing) and decrease in polymer backbone mobility and (ii) weakening interchain interactions to avoid electronic interactions. The introduction of rigid and bulky packing-disrupting units into the backbone has been proposed as the most effective way of enhancing diffusivity and selectivity through hindrance of packing of the relatively rigid backbones and reduction of the rotational mobility around the flexible groups.15–19
A large number of polymeric materials including polyimides,20,21 poly(amide–imide)s,22 polybenzimidazoles,23 polycarbonates,24 and polysulfones25 have been investigated and developed as promising membranes for CO2 separation applications. However, these membranes were found to develop shortcomings including limited thermal and chemical stability together with suffering from the usual trade-off between the desirable properties of selectivity and permeability.
Thermally stable and chemically resistant heteroaromatic polymers such as polyoxadiazoles (PODs) and polyhydantoins have been utilized to prepare dense membranes for CO2 separation since 1994.26–29 Despite their excellent thermal and chemical stabilities and good mechanical properties complemented by high selectivity and good permeability, application of POD membranes in separation technologies were limited by their quite difficult processing and fabrication. Similar to other aromatic rigid polymers, these fully aromatic backbones are not soluble in any organic solvents and exhibit a very high (or no) glass transition temperature. Various attempts have been made to modify the solubility and processability of the rigid chain polymers by introducing either noncoplanar or bulky groups,30–32 groups with greater rotational freedom such as –O–, –CH2–, –C(CH3)2– and –C(CF3)2–,33–36 or less symmetric units such as ortho- and meta-catenated aromatic rings in the main chains.37–39
Naphthalene is a bulky, rigid and heat resistance moiety.40–42 As observed in our previous studies, the incorporation of ortho-linked binaphthyl-based systems such as 1,1′-thiobis(2-naphthoxy) (S-BINOL) catenate could disrupt the crystal packing, reducing intermolecular interactions and enhancing solubility of the resulting polymers.43,44 Considering such observation, it was envisaged that the introduction of the S-BINOL catenate to the polyoxadiazole backbone might lead to simultaneous increase in the fractional free volume (FFV) and the rigidity of the polymer. The former leads to higher diffusivity whereas, the latter results in significant selectivity improvement.
In the present study, in order to prepare efficient membranes, relatively rigid polyoxadiazole backbones containing poor chain packing S-BINOL and SO2-BINOL catenates were designed and synthesized. The general characteristics of the obtained polymers such as crystallinity, thermal and physical properties were investigated. The performance of the membranes with respect to CO2 separation under various feed compositions, pressures, and temperatures were also evaluated and presented. A special attention was also given to establish a correlation between the structural variations in the polymer backbone and their impact on the gas transport properties.
Experimental
Materials
1,1′-Thiobis(2-naphthol) (S-BINOL) and 1,1′-thiobis(2-naphthoxy acetic ester) (S-BINOL-DE) were synthesized according to the procedure reported in a previous communication.44 Hydrazine monohydrate (Acros), terephthaloyl chloride (Merck) and polyphosphoric acid (PPA, Merck) were used as received. N-methyl-2-pyrrolidone (NMP, Merck) and pyridine (Py, Fluka) were purified by distillation under reduced pressure over calcium hydride and stored over 4 Å molecular sieves. LiCl (Merck) was dried under vacuum at 180 °C before use.Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N–H)), 8.39 (d, 2H, J = 8.4 Hz, Ar H), 7.85 (d, 2H, J = 9.1 Hz, Ar H), 7.82 (d, 2H, J = 8.1 Hz, Ar H), 7.41 (d, 2H, J = 8.3 Hz, Ar H), 7.33 (d, 2H, J = 9.2 Hz, Ar H), 7.32 (d, 2H, J = 8.2 Hz, Ar H), 4.57 (s, 4H, CH2), 4.33 (s, 4H, NH2); 13C NMR (75 MHz, DMSO-d6, δ): 166.34 (CO), 158.24 (Ar), 135.48 (Ar), 130.32 (Ar), 129.60 (Ar), 128.24 (Ar), 127.34 (Ar), 125.81 (Ar), 123.24 (Ar), 120.02 (Ar), 115.14 (Ar), 65.71 (CH2); IR (KBr): ν = 3306 (s, N–H stretching), 3045 (w, Ar-H stretching), 2925 (w, C–H stretching), 1670 hydrazide I band (s, hydrazide CO stretching), 1620 hydrazide II band (m, N–H bending), 1530 (w), 1505 (m), 1305 (w), 1265 (s), 1230 (w), 1150 (w), 1075 (w), 1000 (w), 915 (w), 805 (m), 800 (m), 695 (w), 616 (w) cm−1. Anal. calcd for C24H22O4N4S (%): C 62.32, H 4.79, N 12.11; found, C 62.27, H 4.69, N 11.81.O stretching), 1620 hydrazide II band (m, N–H bending), 1590 (w), 1500 (m), 1460 (w), 1260 (s), 1225 (s), 1150 (w), 1080 (m), 810 (m), 760 (w) cm−1. Anal. calcd for C32H24N4O6S (%): C 64.85, H 4.08, N 9.45; found, C 64.09, H 4.58, N 9.10.
C–N–H)), 8.39 (d, 2H, J = 8.4 Hz, Ar H), 7.85 (d, 2H, J = 9.1 Hz, Ar H), 7.82 (d, 2H, J = 8.1 Hz, Ar H), 7.41 (d, 2H, J = 8.3 Hz, Ar H), 7.33 (d, 2H, J = 9.2 Hz, Ar H), 7.32 (d, 2H, J = 8.2 Hz, Ar H), 4.57 (s, 4H, CH2), 4.33 (s, 4H, NH2); 13C NMR (75 MHz, DMSO-d6, δ): 166.34 (CO), 158.24 (Ar), 135.48 (Ar), 130.32 (Ar), 129.60 (Ar), 128.24 (Ar), 127.34 (Ar), 125.81 (Ar), 123.24 (Ar), 120.02 (Ar), 115.14 (Ar), 65.71 (CH2); IR (KBr): ν = 3306 (s, N–H stretching), 3045 (w, Ar-H stretching), 2925 (w, C–H stretching), 1670 hydrazide I band (s, hydrazide CO stretching), 1620 hydrazide II band (m, N–H bending), 1530 (w), 1505 (m), 1305 (w), 1265 (s), 1230 (w), 1150 (w), 1075 (w), 1000 (w), 915 (w), 805 (m), 800 (m), 695 (w), 616 (w) cm−1. Anal. calcd for C24H22O4N4S (%): C 62.32, H 4.79, N 12.11; found, C 62.27, H 4.69, N 11.81.O stretching), 1620 hydrazide II band (m, N–H bending), 1590 (w), 1500 (m), 1460 (w), 1260 (s), 1225 (s), 1150 (w), 1080 (m), 810 (m), 760 (w) cm−1. Anal. calcd for C32H24N4O6S (%): C 64.85, H 4.08, N 9.45; found, C 64.09, H 4.58, N 9.10.Cyclodehydration of polyhydrazides
Both thermal and chemical cyclodehydration methods were used to convert polyhydrazides into corresponding polyoxadiazoles of t-POD or t-OPOD and s-POD, respectively. In the first case, the reactions were performed on films under vacuum while in the chemical method, solution of PPA was used for cyclodehydration in an inert atmosphere.t-POD. Inherent viscosity: 1.06 dL g−1; cyclization yield: 95.6% (based on thermal analysis data); IR: ν = 3205 (w), 3050 (w, Ar-H stretching), 2940, 1605, 1580 (m, C
N stretching), 1525 (w), 1490 (w), 1230 (m), 1175 (w), 1040 (m), 955 (w), 815 (w), 750 (w) cm−1. Anal. calcd for C32H20N4O4S (%): C 69.05, H 3.62, N 10.07; found, C 68.31, H 3.81, N 9.70.
t-OPOD. Cyclization yield: 95.2% (based on thermal analysis data); IR: ν = 3100 (w), 3065 (w, Ar-H stretching), 2930 (w, C–H stretching), 1600 (m), 1580 (m, C
N stretching), 1490 (w), 1315 (w), 1230 (m), 950 (w), 825 (m), 750 (m), 720 (w) cm−1. Anal. calcd for C32H20N4O6S (%): C 65.30, H 3.45, N 9.50; found, C 64.78, H 3.92, N 9.16%.
N stretching), 1525 (w), 1495 (w), 1235 (m), 1170 (w), 1040 (w), 1010 (m), 955 (w), 815 (m), 750 (w), 715 (w) cm−1. Anal. calcd for C32H20N4O4S (%): C 69.05, H 3.62, N 10.07; found, C 68.27, H 3.73, N 9.60%.Oxidation of sulfide to sulfone
s-OPOD. Inherent viscosity: 0.93 dL g−1; IR (KBr): ν = 3105 (w), 3060 (w, Ar-H stretching), 2920 (w), 1585 (m, C
N stretching), 1580 (m), 1490 (w), 1315 (m), 1235 (m), 950 (m), 750 (m), 725 (w) cm−1. Anal. calcd for C32H20N4O6S (%): C 65.30, H 3.45, N 9.50; found, C 64.87, H 3.85, N 9.07%.
OPH. Inherent viscosity: 0.98 dL g−1; IR (KBr): ν = 3295 (s, N–H stretching), 3050 (w, Ar-H stretching), 1675 hydrazide I band (s, hydrazide C
O stretching), 1620 hydrazide II band (m, N–H bending), 1595 (w), 1490 (m), 1460 (m), 1317 (w), 1255 (s), 1225 (s), 1080 (m), 810 (m) cm−1. Anal. calcd for C32H24N4O8S (%): C 61.53, H 3.87, N 8.97; found, C 63.17, H 4.02, N 8.60%.
Characterization techniques
IR spectra were recorded in potassium bromide (KBr) pellets or films on a Perkin Elmer (Perkin Elmer FT spectrum RX1, USA) over the range 400–4000 cm−1. 1H NMR and 13C NMR spectra were recorded using Bruker DRX AVANCE 300. Elemental analysis performed by a Perkin Elmer 2004 (II) CHN analyzer. Melting points (uncorrected) were measured with an Electrothermal Engineering LTD 9200 apparatus. Inherent viscosities ( at a concentration of 0.5 g dL−1) were measured with an Ubbelohde suspended-level viscometer at 30 °C using NMP as solvent. Thermogravimetric analysis were recorded on a thermal analyzer 931 TA instrument under nitrogen atmosphere at a heating rate of 10 °C min−1. Glass-transition temperatures were measured on a differential scanning calorimetry (DSC) 950 TA analyzer with a heating rate of 10 °C min−1. Tensile strength and elongation at break of thin films were evaluated with a Shimadzu AG-10-TB at room temperature and in 5% strain rate of the sample specimen length. Wide angle X-ray diffraction measurements were recorded at room temperature on a PW 1800 (Philips) over a range of 2θ = 4–80°. Based on the Bragg's law, the average d-spacing (d) was calculated as following
at a concentration of 0.5 g dL−1) were measured with an Ubbelohde suspended-level viscometer at 30 °C using NMP as solvent. Thermogravimetric analysis were recorded on a thermal analyzer 931 TA instrument under nitrogen atmosphere at a heating rate of 10 °C min−1. Glass-transition temperatures were measured on a differential scanning calorimetry (DSC) 950 TA analyzer with a heating rate of 10 °C min−1. Tensile strength and elongation at break of thin films were evaluated with a Shimadzu AG-10-TB at room temperature and in 5% strain rate of the sample specimen length. Wide angle X-ray diffraction measurements were recorded at room temperature on a PW 1800 (Philips) over a range of 2θ = 4–80°. Based on the Bragg's law, the average d-spacing (d) was calculated as following
 | (1) |
The film density was determined in a Ca(NO3)2 solution with standard buoyancy method. The following equation was used to calculate the density value.
 | (2) |
 | (3) |
Gas separation measurements
The pure and mixed gas permeability coefficients were measured using a constant volume, variable pressure apparatus at different feed pressures and temperatures as described elsewhere.48 A gas chromatography (8700 Perkin Elmer) equipped with a thermal conductivity detector (GC-TCD) was used to evaluate the concentration of each gas in the permeate side. The permeability of each gas (Pi) was described in Barrer (cm3 (STP) cm−2 s−1 cmHg−1) by the following relationship:
 | (4) |
 , which is defined as the ratio of pure gas permeability of each species.
, which is defined as the ratio of pure gas permeability of each species.
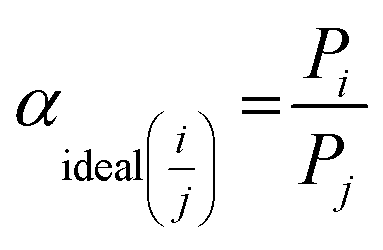 | (5) |
The real selectivity of membranes was calculated with mixed gas as follows:
 | (6) |
Results and discussion
Monomer and polymer synthesis and characterization
The monomer was synthesized in a high purity form S-BINOL by a two-step high-yielding procedure as shown in Fig. 1. Firstly, the treatment of S-BINOL with methylcholoroacetate gave corresponding diester in 90% yields. Subsequently, the dihydrazide monomer was readily obtained by refluxing of the intermediate diester with the excess of hydrazine hydrate. | ||
| Fig. 1 Monomer synthesis. | ||
The structure of the S-BINOL-DH monomer was confirmed by FT-IR, 1H and 13C NMR, and elemental analysis. Assignments of each proton are assisted in 1H spectrum and shown in Fig. 2. As could be seen, the data agreed well with the proposed structure of the synthesized monomer. Two resonance signals at most downfield and upfield regions; 9.02 (s, 2H, NH protons) and 4.3 (s, 4H, NH2 protons) ppm are ascribed to the protons of hydrazide groups (Hi and Ha). Also, the area of integration for protons is in accordance with the assignments.
 | ||
| Fig. 2 1H NMR spectrum of S-BINOL-DH in DMSO-d6. | ||
Polyhydrazides containing S-BINOL were synthesized by polycondensation method as described in Fig. 3. In addition to terephthaloyl chloride monomer, the polymerization of the isophthaloyl chloride with S-BINOL-DH was also attempted under various conditions by changing the reaction temperature and time. However, these attempts were not successful and the resulting low molecular weight polymers produced brittle films or films with low mechanical stability. Hence, terephthaloyl chloride based materials were alternatively used for the next experiments.
 | ||
| Fig. 3 Synthesis of the polyhydrazides, cyclodehydration, and oxidation to the corresponding sulfone containing polymers. | ||
Polyoxadiazoles were prepared by cyclodehydration of polyhydrazide films by thermal treatment or solution method using polyphosphoric acid as a dehydrating agent, according to the route depicted in Fig. 3.
Upon cyclodehydration, the intensive absorption peaks at around 3200–3300 cm−1 due to the N–H stretching and 1670 cm−1 arising from carbonyl groups of polyhydrazide were almost disappeared. In addition, the characteristic bands of the oxadiazole ring at 1580, 1010 and 950 cm−1 were appeared (Fig. 4). Dehydration to the oxadiazole structure was also confirmed by the CHN elemental analysis. The carbon mass showed more than 4% weight increase due to the dehydration and C 68.27 wt%, H 3.73 wt% and N 9.60 wt% (s-POD) is well consistent with the calculated values of C 69.05 wt%, H 3.62 wt% and N 10.07 wt%. The slightly higher contents of hydrogen in CHN data and small peaks above 3200 cm−1 in IR can be attributed to the absorbed water or incomplete cyclodehydration.
 | ||
| Fig. 4 FT-IR spectrum of S-BINOL-DH, corresponding polyhydrazide (PH) and chemically cyclodehydrated polyoxadiazole (POD). | ||
Since the hydrazide group is considered to be less stable than oxadiazole ring, thermogravimetric data can be used to evaluate the degree of cyclization reaction or the hydrazide content (%HZ).49 It was determined from the weight loss of residual hydrazide groups of polyoxadiazoles in distinct regions of temperatures between 270 and 325 °C. The following equation was used to calculate the hydrazine content,
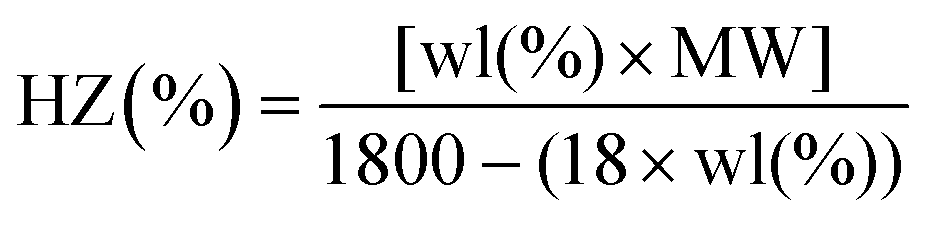 | (7) |
 | ||
| Fig. 5 Thermogravimetric analysis of PH, s-POD and s-OPOD (top) and expanded regions of distinct weight losses in PODs (down) under N2 atmosphere at a heating rate of 10 °C min−1. | ||
Finally, the sulfide group of polyoxadiazole and polyhydrazide readily oxidized into sulfone by means of hydrogen peroxide. Although the symmetric stretching band of sulfone group is not a good indicator for this oxidation due to its overlap with other absorption bands, the asymmetric band at around 1315 cm−1 clearly confirms the oxidation. In addition, elemental analysis shows good agreement with the expected values for oxidation. Based on the agreement between calculated and found data, introducing of sulfone group through oxidation of polyoxadiazole is preferred to oxidation of polyhydrazide.
X-ray diffraction and solubility of the polymers
Wide-angle X-ray diffraction patterns were used to investigate the morphology of the polyoxadiazoles and morphological differences between the sulfide and sulfone bridged arrangements. As shown in Fig. 6, the curves of both polyoxadiazoles were broad and without obvious peak features, indicating their amorphous structure. This amorphous nature can be mainly attributed to the presence of the noncoplanar ortho-linked structure in the polymer backbone which hinder efficient chain packing and decrease the regularity through weakening intermolecular forces between the polymer chains; subsequently causing a decrease in crystallinity. | ||
| Fig. 6 WAXD patterns of s-OPOD (top) and s-POD membranes. | ||
The structural noncoplanarity and cisoid conformation of the two naphtyl rings is also evident from the geometry of the S-BINOL monomer; optimized with MM2 method using the ChemBio3D Ultra 11.0, shown in Fig. 7 and X-ray structure.42,50
 | ||
| Fig. 7 Geometry optimized structure of the S-BINOL monomer. | ||
The WAXD data could also be used to calculate the average d-spacing of a polymer matrix according to the eqn (4) (Table 1). The higher d-spacing value means more intermolecular distances and therefore more easily passing of penetrant molecules. Since the bridged sulfone group in the ortho-position causes more steric repulsion between two naphtyl groups on BINOL catenate, the rotational energy barrier around this bond will further increase (shows higher FFV). The XRD patterns in Fig. 6 and d-spacing values in Table 1 support the idea of decreased chain stacking resulting from oxidation of sulfide atom in the polymer backbone. This decreased planarity along with the polymer backbone would increase FFV and facilitate the gas permeation through the membrane.
| Polymer | Mechanical properties | d-spacing | Vw | ρ | FFV | P (barrers) | αideal
|
||
|---|---|---|---|---|---|---|---|---|---|
| Tensile strength (MPa) | Elongation at break (%) | (Å) | cm3 g−1 | g cm−3 | CO2 | CH4 | |||
| PH | 32.77 | 8.15 | NA | NA | NA | NA | NA | NA | NA |
| OPH | 30.46 | 7.94 | NA | NA | NA | NA | NA | NA | NA |
| s-POD | 21.16 | 5.46 | 4.28 | 0.538 | 1.216 | 0.150 | 8.04 | 0.09 | 89.33 |
| t-POD | 8.42 | 2.60 | 4.25 | 0.538 | 1.215 | 0.151 | 9.63 | 0.18 | 53.50 |
| s-OPOD | 20.68 | 4.21 | 5.13 | 0.547 | 1.137 | 0.192 | 18.75 | 0.26 | 72.11 |
| t-OPOD | 8.01 | 2.67 | 5.06 | 0.547 | 1.137 | 0.192 | 15.52 | 0.31 | 50.06 |

As shown in Fig. 6, the POD shows a maximum peak at about 2θ = 20.7°, corresponding to an average inter-segmental distance of 4.28 Å; after oxidation, this peak shifts to the left to 2θ = 17.6°, suggesting the presence of a higher average distance around 0.85 Å in OPOD. Meanwhile, the OPOD shows a small broad shoulder on the high-angel side at 37° corresponding to a d-spacing of 2.42 Å. Based on the similar observations for polyimides,51–53 this halo could be assigned to π–π stacking of oxadiazole and naphthyl rings in ordered domains. Existing of such stacking in sulfone containing polymers confirm the more rigidity of their backbones than corresponding sulfide bridged analogues.
Solubility of the high performance polymers is a critical factor that exhibits its processability for diverse applications. The solubility behavior of these amorphous polyhydrazides and polyoxadiazoles was tested and the obtained data was reported in Table 2. The polymers were found to be soluble in highly polar solvents such as NMP, DMAc, and DMSO, at room temperature or upon heating. Such good solubility behavior was probably attributed to the disturbed dense chain packing of the polymer chain by the bulky S-BINOL; consequently, the solvent molecules were able to solubilize the polymer chains. As can be seen, the solubility varied depending on the bridged atoms between aromatic rings. Polyoxadiazoles derived from sulfide bridged monomer showed improved solubility. In fact, the presence of sulfur linkages in the backbone further increased the overall flexibility and this polymer was found to be even partially soluble in DMF.
| Polymer | Viscosityb | Solventc | Thermal stability (°C) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ηinh (dL g−1) | NMP | DMAc | DMSO | DMF | THF | CHCl3 | Tgd | T10e | |
| a Measured at a polymer concentration of 0.05 g mL−1.b Measured at a polymer concentration of 0.5 g dL−1 in NMP solvent at 30 °C.c NMP: N-methyl-2-pyrrolidone; DMAc: N,N-dimethyl acetamide; DMF: N,N-dimethylformamide; DMSO: dimethyl sulfoxide; THF: tetrahydrofuran.d Midpoint temperature of baseline shift on the DSC heating trace.e Temperature at which 10% weight loss occurred under nitrogen atmosphere.f Could not be measured due to the insolubility in the tested solvents.g (++) soluble at room temperature, (+) soluble after heating, (±) partially soluble, (—) insoluble. | |||||||||
| PH | 1.17 | ++ | ++ | ++ | ++ | ± | — | 155 | 295 |
| OPH | 0.98 | ++ | ++ | ++ | ++ | ± | — | 162 | 295 |
| s-POD | 0.98 | ++ | ++ | + | ± | — | — | 245 | 410 |
| t-POD | 1.06 | + | ± | ± | — | — | — | 273 | 400 |
| s-OPOD | 0.93 | ++ | ++ | ± | — | — | — | 274 | 425 |
| t-OPOD | —f | ± | ± | — | — | — | — | 275 | 405 |
Thermal and mechanical properties
The thermal properties of the polymers, which were evaluated by TGA and DSC techniques, are summarized in Table 2. The DSC analysis shows that there is no evidence of crystallization and melting processes. Therefore, the polymers are in an amorphous state and do not reveal any tendency to crystallize, even in the cooling step. The glass transition temperature of polymers was slightly increased after oxidation, which might be caused by the less flexibility of the sulfone linkages.As shown in Fig. 5, the polyoxadiazole membrane exhibited good thermal stability and the temperature for 10% weight loss (T10) was above 400 °C. Therefore, it can be concluded that the introduction of flexible ether and methylene linkages and S-BINOL catenate did not deteriorate significantly the thermal stability of polyoxadiazoles. They exhibited two distinct transition regions of minor weight loss below 400 °C in nitrogen atmosphere. The first transition occurring between 100–130 °C is associated with the loss of adsorbed water in the polymer. The second transition with an approximate weight loss of less than 2% was positioned in the temperature range of 270 and 325 °C and was due to the loss of water produced during the formation of oxadiazole rings of residual hydrazide groups. This was confirmed by detecting major weight loss for polyhydrazide in the same range of temperature (around 300 °C, Fig. 5). The main region of weight loss associated with the degradation of the main chain was above 410 °C for POD. This weight loss temperature was slightly shifted toward higher temperature (above 425 °C) after oxidation.
Table 1 shows the mechanical properties of the membranes; as can be seen, the tensile strength of polyoxadiazoles is above 20 MPa and elongation at break is in the range of 4–6 (%). After converting sulfide to sulfone group, less force per unit area (tensile strength) is required to break the chains apart; it seems that the intermolecular forces between polymer chains decreased in OPOD.
Gas transport properties
The comparison of these data with some recently reported polymeric membranes for gas separation is represented in Robeson plot as depicted in Fig. 8. As shown, the gas separation performances of all membranes are below the current Robeson's upper bound. The polyoxadiazole membranes demonstrated almost higher selectivity and in some cases even better CO2 permeability in comparison to the previously reported polyoxadiazoles26,27 and polyhydantoins29 containing materials. This is coupled with their much higher solubility (as they are soluble in polar aprotic solvents) and therefore better processability than that of previously reported polyoxadiazoles. Upon comparision with pristine and modified polyethersulfones (PESs),48,54 current membranes exhibited higher permeability and higher selectivity. Comparison with some recently reported modified polyimides (PI),56 Matrimides,57,58 poly(benzoxazole-co-imide)s,59 ionic liquids based copolyimides,60 thermally rearranged PI (TR-PI),61 and PI with bulky group18 indicated that the present membranes exhibited good performances in CO2 separations. Particularly, POD membranes showed lower CO2 permeability and higher selectivity than those of previously mentioned imide based polymers.
 | ||
| Fig. 8 Comparison of CO2/CH4 selectivity vs. CO2 permeability of synthesized membranes with some recently important published data including PES and PES–SDS,48 PES–AS,54 cellulose-acetate,55 polyimides and related composite,56 Matrimide,57 brominated Matrimid,58 poly(benzoxazole-co-imide)s,59 ionic liquid based copolyimide,60 thermally rearranged PI,61 PI with bulky group,18 polyhydantoins (PHYs),29 and some previously reported polyoxadiazoles shown by triangle.26,27 | ||
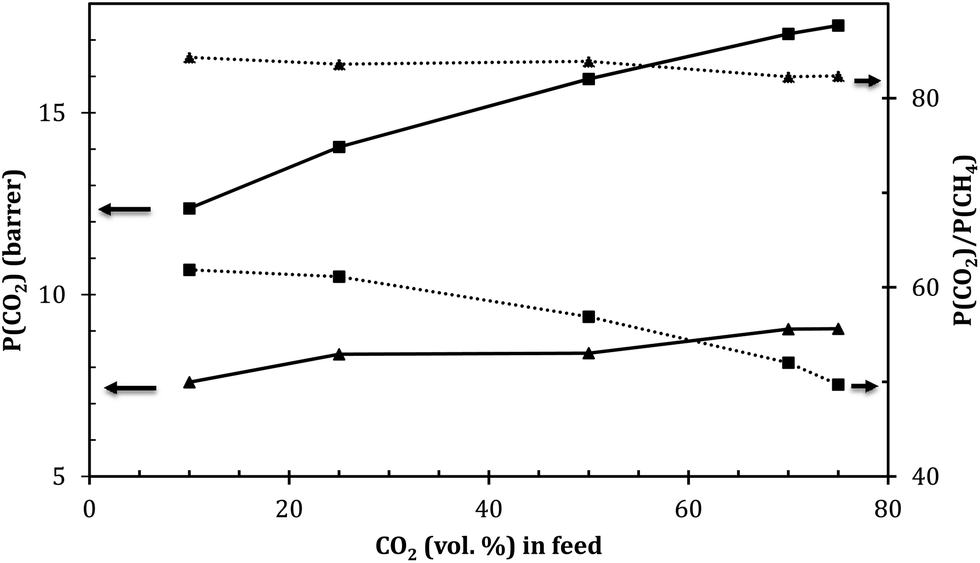 | ||
| Fig. 9 Feed dependence of CO2 permeance and CO2/CH4 selectivity of POD (▲) and OPOD (■). Test conditions: 9 bar feed pressure and 30 °C. | ||
 | ||
| Fig. 10 Temperature dependence of CO2 permeance and CO2/CH4 selectivity of POD (▲) and OPOD (■). Test conditions: 9 bar feed pressure, 10/90 CO2/CH4. | ||
 | ||
| Fig. 11 Effect of feed pressure on CO2 permeance and CO2/CH4 selectivity of POD (▲) and OPOD (■) membranes. Test conditions: 50/50 CO2/CH4, 30 °C. | ||
As seen in Fig. 9, the increase in CO2 volume percent in the feed led to a linear increase in the permeability of CO2. An opposite behavior was observed in selectivity as higher CO2 concentrations in the feed caused appreciably lower selectivity values. Such observed selectivity is due to the increase in the plasticization effect of CO2 with increasing its concentration in the feed and consequent larger free volume of membranes.
As could be seen, the slope of this change is different in both membranes. While the sulfide-containing membrane shows negligible increase in the permeability and decrease in the selectivity with the increase in CO2 concentration in the feed, the sulfone-containing membranes revealed sharper changes with the variation in the feed composition. For instance, when the CO2 concentration in the feed increased from 25 to 50%, the CO2 and CH4 permeabilities increased by 22 and 43% in OPOD and 8.25 and 10% in POD, respectively. This was accompanied by a decrease in the selectivity by 15% in the sulfone-containing membranes (OPOD) compared to 1.6% in POD membranes.
To investigate the temperature dependency of permselectivity, the effect of feed temperature on selectivity and permeability of membranes were investigated. The effect of temperature on the gas permselectivity can be generally understood by considering the temperature dependency of gas diffusivity and solubility. The diffusion of gas molecules can be described by the Arrhenius equation.62 Since the activation energy of the diffusion is mostly positive in glassy polymers, the increase of the feed temperature always increases the gas diffusivity.54 This activation energy (which is an amount of required energy to sufficiently separate polymer chains to allow penetration) is directly related to penetrant size. For example, in gases with higher diffusion diameters such as CH4 (3.817 Å), higher activation energy is needed compared with the gases with lower diffusion diameters such as CO2 (3.325 Å).62 On the other hand, gas solubility in glassy polymers is directly proportional to the Lennard–Jones temperature of the gas and is fairly insensitive to the chemical structure of the polymer, as suggested by Van Krevelen.63 Taking the Lennard–Jones temperature into consideration, CO2 has higher selectivity than CH4.
As shown in Fig. 10, the increase in the temperature in the range of 25–45 °C caused an increase in the gas permeance in both membranes which is in accordance with the above theoretical discussion. Since CH4 has higher activation energy than CO2 and the polymer chains exhibit more mobility in the higher temperatures, the increase in the feed temperature increases permeability of CH4 more than that of CO2, resulting in lower selectivity upon increase in the temperature.
An increase in feed pressure can entail two competing effects in glassy polymers: a decrease in the gas permeability due to the saturation of Langmuir sites and an increase in the gas permeability at plasticization pressure.56 Membrane plasticization is an undesired process usually observed at high feed partial pressure, which increases the permeance and significantly reduces the selectivity. Fig. 11 shows the permeability and selectivity of mixed CO2/CH4 gases (50/50) as a function of feed pressure. As can be seen, the increase in the feed pressure up to 15 bar led to a decrease in the permeability coupled with an increase in the selectivity. Both membranes show some degrees of plasticization phenomenon at a CO2 feed pressure of above 15 bar. It can be concluded that the oxidated (sulfone-containing) membrane displays enhanced plasticization resistance against CO2, possibly due to the lower degree of chain mobility.
Conclusions
Novel dihydrazide monomer containing an ortho-catenated non-coplanar S-BINOL was designed, synthesized, and used as a new building block of polyoxadiazole membranes with high FFV. The incorporation of less symmetric ortho-linked and bulky nature of S-BINOL reduced the polymer backbone symmetry, regularity, and chain packing efficiency which resulted in substantially improved solubility in polar aprotic solvents, decreased crystallinity and high FFV. The thermal stability showed T10 at above 400 °C and membrane recorded a respective ideal selectivity of more than 82% and permeances of 8.04 and 0.09 (barrer) for CO2 and CH4, respectively. Oxidation of the sulfide linkage caused a remarkably improved CO2 permeability (around 18.75 barrer) and lower CO2/CH4 selectivity (around 72%). These changes are in accordance with the calculated data for free volume available for penetrating gas molecules (d-spacing) and local available space in a dense polymer chains (FFV); oxidation caused a significant increase in the FFV and d-spacing of the polymer. In performance evaluation with binary mixed gases, the oxidated membrane displayed an enhanced plasticization resistance against CO2 and its performance was more sensitive to the change in the feed pressure and composition. Even though these new polyoxadiazole membranes demonstrate better performance than the commercial and modified polyethersulfones and previously reported polyoxadiazoles, the permeability of these membranes still remains lower than that of thermally rearranged (TR) polymers. By the way, their remarkably higher selectivity and inherent stability opens important approach for further improvement in gas separation properties of the polymeric membranes.Notes and references
- A. Koltuniewicz, in Comprehensive Membrane Science and Engineering, ed. D. Enrico and G. Lidietta, Elsevier, Oxford, 2010, pp. 109–164 Search PubMed.
- M. G. Buonomenna, RSC Adv., 2013, 3, 5694–5740 RSC.
- D. D. Iarikov and S. Ted Oyama, in Membrane Science and Technology, eds. S. T. Oyama and M. S.-W. Susan, Elsevier, 2011, vol. 14, pp. 91–115 Search PubMed.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- D. Freeman Benny and I. Pinnau, in Advanced Materials for Membrane Separations, American Chemical Society, 2004, vol. 876, ch. 1, pp. 1–23 Search PubMed.
- P. Bernardo, E. Drioli and G. Golemme, Ind. Eng. Chem. Res., 2009, 48, 4638–4663 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS PubMed.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS PubMed.
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS.
- M. Langsam and W. F. Burgoyne, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 909–921 CrossRef CAS.
- W. J. Koros and D. R. B. Walker, Polym. J., 1991, 23, 481–490 CrossRef CAS.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS PubMed.
- J. K. Adewole, A. L. Ahmad, S. Ismail and C. P. Leo, Int. J. Greenhouse Gas Control, 2013, 17, 46–65 CrossRef CAS PubMed.
- S. K. Sen and S. Banerjee, J. Membr. Sci., 2010, 365, 329–340 CrossRef CAS PubMed.
- C. Y. Soo, H. J. Jo, Y. M. Lee, J. R. Quay and M. K. Murphy, J. Membr. Sci., 2013, 444, 365–377 CrossRef CAS PubMed.
- Z. Liu, B. Liu, L. Li, Y. Zhang and Z. Jiang, Macromol. Res., 2013, 21, 608–613 CrossRef CAS.
- M. Calle, C. García, A. E. Lozano, J. G. de la Campa, J. de Abajo and C. Álvarez, J. Membr. Sci., 2013, 434, 121–129 CrossRef CAS PubMed.
- P. Bandyopadhyay, D. Bera and S. Banerjee, Sep. Purif. Technol., 2013, 104, 138–149 CrossRef CAS PubMed.
- S. Sridhar, R. S. Veerapur, M. B. Patil, K. B. Gudasi and T. M. Aminabhavi, J. Appl. Polym. Sci., 2007, 106, 1585–1594 CrossRef CAS.
- S. Kumar Sen and S. Banerjee, RSC Adv., 2012, 2, 6274–6289 RSC.
- P. Bandyopadhyay, D. Bera, S. Ghosh and S. Banerjee, J. Membr. Sci., 2013, 447, 413–423 CrossRef CAS PubMed.
- S. C. Kumbharkar, P. B. Karadkar and U. K. Kharul, J. Membr. Sci., 2006, 286, 161–169 CrossRef CAS PubMed.
- M. Aguilar-Vega and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 1599–1610 CrossRef CAS.
- K. J. Lee, J. Y. Jho, Y. S. Kang, J. Won, Y. Dai, G. P. Robertson and M. D. Guiver, J. Membr. Sci., 2003, 223, 1–10 CrossRef CAS.
- E. R. Hensema, M. E. R. Borges-Sena, M. H. V. Mulder and C. A. Smolders, Gas Sep. Purif., 1994, 8, 149–160 CrossRef CAS.
- M. E. Sena and C. T. Andrade, Polym. Bull., 1995, 34, 643–648 CrossRef CAS.
- C. H. Jung, J. E. Lee, S. H. Han, H. B. Park and Y. M. Lee, J. Membr. Sci., 2010, 350, 301–309 CrossRef CAS PubMed.
- R. Tejero, Á. E. Lozano, C. Álvarez and J. de Abajo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4052–4060 CrossRef CAS.
- E. Abouzari-Lotf, A. Shockravi and A. Javadi, Polym. Degrad. Stab., 2011, 96, 1022–1028 CrossRef CAS PubMed.
- A. Shockravi, E. Abouzari-Lotf and A. Javadi, Des. Monomers Polym., 2009, 12, 119–128 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, Q. Lan, Z. Qin, S. Liu, C. Zhao, Z. Chi and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1302–1314 CrossRef CAS.
- A. Shockravi, E. Abouzari-lotf, A. Javadi and S. Taheri, Polym. J., 2009, 41, 174–180 CrossRef CAS.
- Y. Zhang, P. Luo, H. Yao and S. Guan, React. Funct. Polym., 2012, 72, 621–626 CrossRef CAS PubMed.
- J. Abajo and J. G. Campa, in Progress in Polyimide Chemistry I, ed. H. R. Kricheldorf, Springer, Berlin Heidelberg, 1999, vol. 140, ch. 2, pp. 23–59 Search PubMed.
- A. Ghosh, S. K. Sen, S. Banerjee and B. Voit, RSC Adv., 2012, 2, 5900–5926 RSC.
- A. Shockravi, S. Mehdipour-Ataei, E. Abouzari-Lotf and A. Yousefi, Eur. Polym. J., 2006, 42, 133–139 CrossRef CAS PubMed.
- J. M. García, F. C. García, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623–686 CrossRef PubMed.
- H. Kiani, M. Nasef, A. Javadi, E. Abouzari-Lotf and F. Nemati, J. Polym. Res., 2013, 20, 1–12 CAS.
- J. Ghim, H.-S. Shim, B. G. Shin, J.-H. Park, J.-T. Hwang, C. Chun, S.-H. Oh, J.-J. Kim and D.-Y. Kim, Macromolecules, 2005, 38, 8278–8284 CrossRef CAS.
- D.-J. Liaw, F.-C. Chang, M.-k. Leung, M.-Y. Chou and K. Muellen, Macromolecules, 2005, 38, 4024–4029 CrossRef CAS.
- A. Shockravi, A. Javadi and E. Abouzari-Lotf, RSC Adv., 2013, 3, 6717–6746 RSC.
- A. Shockravi, A. Javadi and E. Abouzari-Lotf, Polym. J., 2011, 43, 816–825 CrossRef CAS.
- A. Shockravi, S. Mehdipour-Ataei, E. Abouzari-Lotf and M. Zakeri, Eur. Polym. J., 2007, 43, 620–627 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- Hypercube Inc., Gainesville, Florida, version 7.0 edn, 2002.
- A. Gavezzotti, J. Am. Chem. Soc., 1983, 105, 5220–5225 CrossRef CAS.
- S. Saedi, S. S. Madaeni, A. Arabi Shamsabadi and F. Mottaghi, Sep. Purif. Technol., 2012, 99, 104–119 CrossRef CAS PubMed.
- D. Gomes, J. C. Pinto and C. Borges, Polymer, 2003, 44, 6223–6233 CrossRef CAS.
- J. T. Mague, M. S. Balakrishna, B. Punji and D. Suresh, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o487–o488 CAS.
- R. Pan, T. Zhou, A. Zhang, W. Zhao and Y. Gu, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 2257–2261 CrossRef CAS.
- M. Calle, A. E. Lozano, J. de Abajo, J. G. de la Campa and C. Álvarez, J. Membr. Sci., 2010, 365, 145–153 CrossRef CAS PubMed.
- J. Wakita, S. Jin, T. J. Shin, M. Ree and S. Ando, Macromolecules, 2010, 43, 1930–1941 CrossRef CAS.
- S. Saedi, S. S. Madaeni, F. Seidi, A. A. Shamsabadi and S. Laki, Int. J. Greenhouse Gas Control, 2013, 19, 126–137 CrossRef CAS PubMed.
- A. C. Puleo, D. R. Paul and S. S. Kelley, J. Membr. Sci., 1989, 47, 301–332 CrossRef CAS.
- M. Askari and T.-S. Chung, J. Membr. Sci., 2013, 444, 173–183 CrossRef CAS PubMed.
- T. T. Moore and W. J. Koros, J. Appl. Polym. Sci., 2007, 104, 4053–4059 CrossRef CAS.
- M. D. Guiver, G. P. Robertson, Y. Dai, F. Bilodeau, Y. S. Kang, K. J. Lee, J. Y. Jho and J. Won, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4193–4204 CrossRef CAS.
- C. A. Scholes, C. P. Ribeiro, S. E. Kentish and B. D. Freeman, J. Membr. Sci., 2014, 450, 72–80 CrossRef CAS PubMed.
- P. Li and M. R. Coleman, Eur. Polym. J., 2013, 49, 482–491 CrossRef CAS PubMed.
- D. F. Sanders, Z. P. Smith, C. P. Ribeiro Jr, R. Guo, J. E. McGrath, D. R. Paul and B. D. Freeman, J. Membr. Sci., 2012, 409–410, 232–241 CrossRef CAS PubMed.
- B. W. Rowe, L. M. Robeson, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2010, 360, 58–69 CrossRef CAS PubMed.
- D. W. Van Krevelen and K. Te Nijenhuis, in Properties of Polymers, Elsevier, Amsterdam, 4th edn, 2009, pp. 655–702 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |