Multifunctional linker for orthogonal decoration of gold nanoparticles with DNA and protein†
Dania M. Kendziora,
Ishtiaq Ahmed and
Ljiljana Fruk*
Centre for Functional Nanostructures, Karlsruhe Institute of Technology (KIT), Wolfgang Gaede Street 1a, 76131 Karlsruhe, Germany. E-mail: Ljiljana.Fruk@kit.edu; Fax: +49 721 608 48495; Tel: +49-721-608 45800
First published on 3rd April 2014
Abstract
A novel trifunctional linker was prepared to allow multiple orthogonal functionalization of gold nanoparticles (AuNPs) with biomolecules. Thiooctic groups contained within the linker act as AuNP surface anchors, while two additional orthogonal functional groups are used for attachment of heme and DNA using amide and copper catalyzed Huisgen cycloaddition, respectively. We demonstrate that heme can act as a platform for reconstitution of a functional heme protein such as myoglobin. Using multifunctional linker, fully functional Au–DNA–Mb hybrids were obtained and such multifunctional constructs expand the synthetic toolbox for nanomaterial tailoring and design of biosensors and novel catalytic materials.
Introduction
Due to their unique chemical and physical properties, metallic nanoparticles have found numerous applications in bionanotechnology, in particular in design of biosensing platforms and drug delivery systems.1–4 Most recent developments include the use of AuNPs for gene regulation due to the control of RNAse protein5 or design of vaccines against tumor cells.6 However, all of these applications require bio functionalization of AuNPs with various biomolecules such as DNA, peptides or antibodies.7 One of the most commonly used method to enable the attachment of biomolecules is a thiol–gold surface interaction,8 which has successfully been employed for immobilization of thiolated DNA9 or cystein containing peptides and proteins.10 However, when trying to bind biomolecules onto an inorganic nano core, often the first challenge is a choice of a mild and biocompatible chemical strategy that enables the attachment without compromising the biological activity of the biomolecule or the stability of NPs.7,11,12 Namely, NPs aggregation can occur under biological conditions, which subsequently leads to the loss of inherent properties such as strong surface plasmon13 or hyperthermia effect.14 Despite the advances achieved in attaching different classes of proteins (antibodies, STV, peroxidases, fluorescent proteins) and DNA to the surface of metallic NPs such as Ag, Au and quantum dots, protein attachment still suffers both from little control over the stereochemistry and stoichiometry of the attachment and in some cases, protein deactivation.12,15For development of sensing platforms or drug delivery systems, where i.e. cell binding species need to be attached together with therapeutic agents, multiple modifications of NPs are required. Up till now this has been achieved through the addition of two or more ligands containing different functional groups in various ratios to which desired molecular species can be attached.16,17 However, this often complicates the surface coverage analysis, in particular the determination of the quantity of different species on the surface.18 To achieve multifunctionality, Beltram et al. prepared a linker containing a thiol group for AuNP binding and carboxy- and amino groups for biomolecule attachment, which might lead to cross-linking between amino and carboxy groups of different AuNPs and therefore aggregation of NPs if the conditions are not carefully controlled.19 In general, use of multifunctional linkers for NPs modification is scarce and one example was demonstrated by Graham and coworkers while designing surface resonance Raman scattering (SERRS) probes.20 A SERRS active dye containing AgNP binding group benzotriazole was introduced to the side chains of commercially available polymers featuring an additional carboxyl group for further modification with e.g. amino-modified oligonucleotides. Although this method facilitated the direct labeling of AgNP with SERRS active, NP binding dye and a biomolecule, it is limited to one particular SERRS label and introduction of the new optical labels requires the synthesis of new ligands limiting the practicality of the approach.20
To enable the addition of multiple and structurally different species onto Au NPs using single linker, we designed tri-functional linker containing AuNP anchoring group and two additional functional groups to enable binding of a range of different species (biomolecules, drugs, tags) in various desired combinations. To demonstrate the applicability of such linker, we present proof of concept study where amine and alkyl/azide functional groups of the multifunctional linker are used for Huisgen cycloaddition with DNA and amide coupling of cofactor heme, which can subsequently be used for the immobilization of protein through cofactor reconstitution.21
Results and discussion
Synthesis of orthogonal linker and Au NPs
A one-pot synthesis of AuNPs was previously reported using lipoic acid derivatives containing thioctic moieties for Au binding, which resulted in spherical NPs of varying sizes depending on the ratio between a ligand and a gold precursor.22,25 Therefore, we designed trifunctional linkers in such way to contain thioctic moiety able to bind and stabilize AuNP surface.In addition, such trifunctional linker contains an amine to which different functional moieties can be added through amide coupling and bioorthogonal alkyne or azide groups capable of undergoing the copper-catalyzed Huisgen 1,3-dipolar cycloaddition (Scheme 1).26,27 Namely, Huisgen 1,3-dipolar cycloaddition has already successfully been employed to enable mild and specific attachment of DNA to proteins28 or for covalent crosslinking of Au NPs.29
 | ||
| Scheme 1 Synthesis of trifunctional linkers L1 and L2. Shown is the schematic representation of the organic synthesis of alkyne and azide-modified linkers. (a) EDC–HCl, HOBt, NMM, THF, 12 h, r.t., (b) piperidin, MeCN, 2 h, r.t., (c) lipoic acid, EDC–HCl, HOBt, NMM, 18 h, r.t., (d) TFA, CH2Cl2, 2 h, r.t. | ||
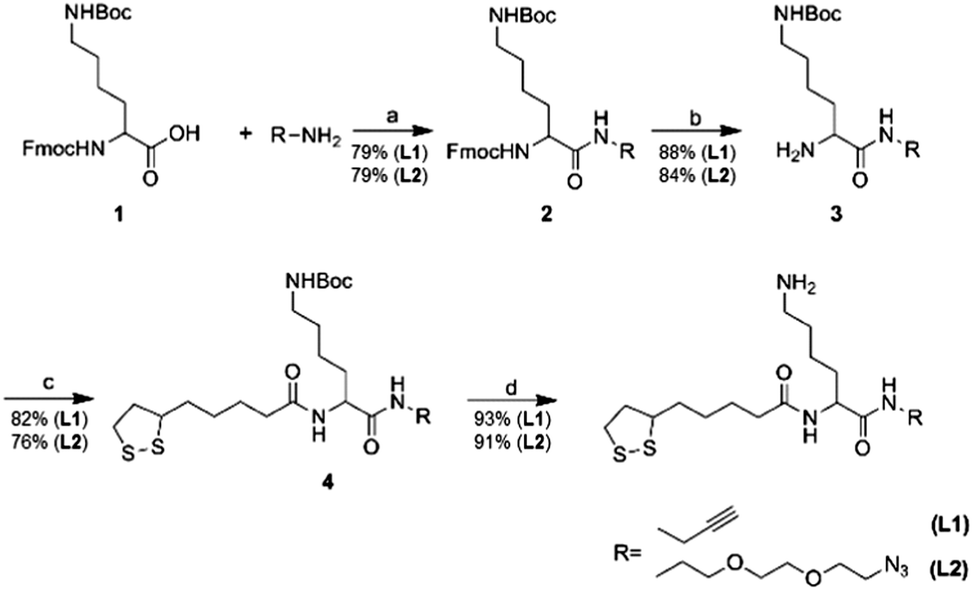
Two different trifunctional linkers L1 and L2 shown in Scheme 1 were synthesized from commercially available Fmoc-Lys(Boc)-OH 1 and were used for preparation of Au NPs. Reactions conditions were first optimized using different ratios of linker to HAuCl4 precursor followed by reduction with NaBH4 (Fig. 1a). Besides alkyne and azide containing linker, unmodified lipoic acid (LA) was used to prepare lipoic acid coated Au NPs to be employed as a control in the subsequent experiments. NP formation was directly assessed by appearance of the plasmon absorption maximum around 520 nm in UV-vis spectra characteristic for smaller AuNPs.30 However, when azide containing linker was used only dark precipitate was formed indicating the precipitation of AuNP aggregates. This is probably due to the reduction of azide to amine during the one pot synthesis of AuNPs in the presence of NaBH4 making it incapable of undergoing subsequent click reactions.31 Therefore only multifunctional linker containing alkyne was used in the subsequent experiments. The most stable and monodisperse 7.4 nm AuNP–L1 were prepared using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 ratio of the linker to precursor as seen from UV-vis and TEM images (Fig. 1b and c) and were further employed for attachment of DNA and heme.
:20 ratio of the linker to precursor as seen from UV-vis and TEM images (Fig. 1b and c) and were further employed for attachment of DNA and heme.
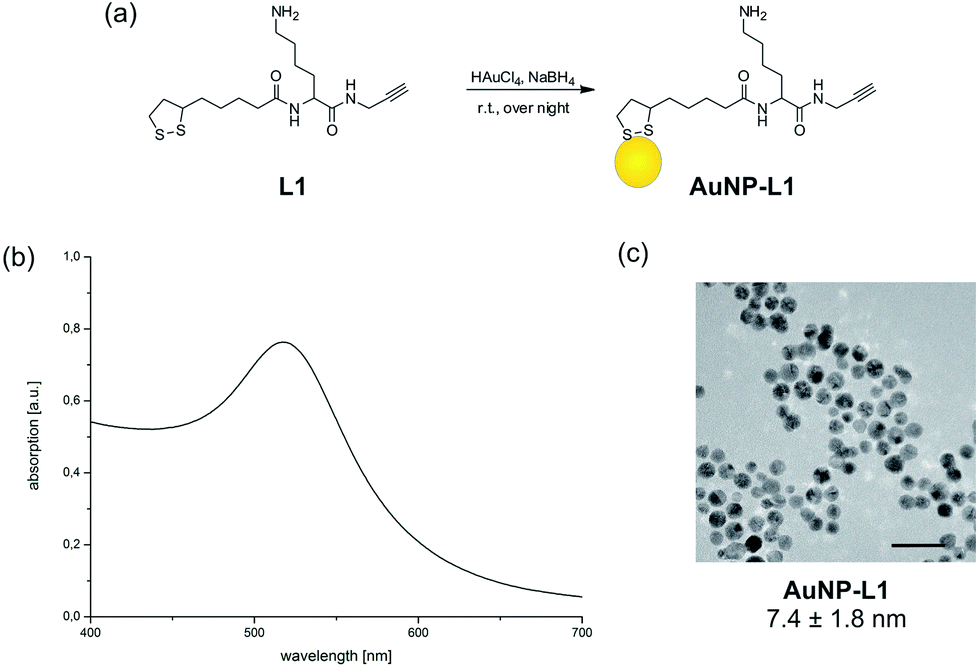 | ||
| Fig. 1 (a) Synthesis of AuNP using trifunctional linker L1. (b) UV-vis spectrum of AuNP–L1 after synthesis using a linker to Au ratio of 1:20. (b) TEM image of AuNP–L1. Scale bar represents 20 nm. | ||
Orthogonal attachment of DNA and heme cofactor
Huisgen dipolar cycloaddition was used to attach azide modified DNA (azDNA) using sodium ascorbate and copper sulfate together with the stabilizing ligand tris(benzyltriazolylmethyl)amine (TBTA) resulting in highly water dispersible DNA-modified AuNPs (AuNP–L1–D). Successful formation of AuNP–L1–D conjugate was confirmed by agarose gel. As shown in Fig. 2, AuNP–L1 precipitated in the gel pocket while AuNP–L1–D migrated within the gel indicating the presence of DNA strands attached to the surface of Au. Negative control in which AuNP–L1 was mixed with azDNA without Cu catalyst at first showed an electrophoretic shift probably due the non specific binding of azDNA, however such NP–DNA conjugates were not stable as the black precipitates were formed after removal of azDNA by centrifugation and washing. | ||
| Fig. 2 1% agarose gel of the modification of AuNP–L1 towards AuNP–D–Mb conjugates. Shown is the electrophoretic separation of different AuNP conjugates after 20 min at 100 V. AuNPs appear as red bands in the gel without additional staining. | ||
To confirm the multifunctionality of the attached L1, AuNP–L1–D was further modified with heme using HOBt/HBTU mediated coupling of the free amine on the AuNP surface and carboxylic groups of the heme molecule (Fig. 3). We have chosen heme for several reasons. First, heme is an important cofactor for a range of enzymes such as metabolically important P450 proteins or peroxidases.32,33 As such, it has already successfully been used to alter existing and introduce new functionalities to these proteins using modified heme reconstitution.21,34,35 Second, heme itself has shown potential to be used as an effective catalyst for a range of reactions and has been utilized for design of artificial enzymatic systems.36,37 After heme coupling, analysis of AuNP–L1–D–heme with UV-vis spectroscopy and comparison of molar concentrations calculated from the specific absorption of heme (405 nm) and AuNP (520 nm) showed that approx. 82 heme molecules per NPs are present.
 | ||
| Fig. 3 Preparation of AuNP–DNA–Mb conjugates using trifunctional linker. Modification of the amine containing DNA–AuNP with heme and subsequent reconstitution with apoMb to yield functional AuNP–L1–D–Mb conjugates. (i) 1000 eq. heme, 1500 eq. HBTU, 2000 eq. HOBt, 3600 eq. DIPEA, 10 min, r.t., (ii) add to 1 eq. AuNP–L1–D, 48 h, r.t., (iii) 1 eq. apoMb, 16 h, r.t. Also shown is the UV-vis spectra of the heme-modified AuNP-DNA before and after reconstitution with apoMb indicating a shift in the Soret band of the heme cofactor upon reconstitution. | ||
Heme reconstitution and Au–DNA-protein functionality
Once heme attachment was confirmed, the Au hybrid was further used for the attachment of myoglobin (Mb) protein using apoMb (Mb without the cofactor) reconstitution. ApoMb was prepared by extraction of native heme using Teale's method24,38 and 8-fold excess of apoMb was added to the heme modified AuNP–L1–D, incubated for 4 hours (Fig. 3) and successful reconstitution was confirmed by UV-vis spectroscopy. As shown in Fig. 3, reconstitution resulted in a shift of heme Soret band absorbance from 390 to Mb specific 406 nm and a change of peak intensity and 520 (AuNP)/Soret band ratio. Using the same methodology as described before, an amount of 53 immobilized Mb was calculated, showing a reconstitution yield of 65%.Successful Mb reconstitution was further confirmed by agarose gel electrophoresis as shown in Fig. 2. As mentioned earlier, agarose gel electrophoresis not only provided an additional proof for the attachment of DNA molecules onto the AuNP surface (high mobility band observed in lane 2, Fig. 2), but also for the coupling of heme (small decrease in mobility in lane 3 as compared to lane 2, Fig. 2). In addition, as seen from lane 4, electrophoretic mobility further decreases upon the reconstitution of apoMb on the NP surface due to the change in the size of the hybrid construct. Apo Mb was employed as a negative control, which is not visible without staining (lane 5). Furthermore, additional control sample containing AuNP–Mb conjugates prepared by direct amide coupling of native Mb to lipoic acid AuNP-0 was added to the gel. As it can be seen in lane 6, such conjugates show electrophoretic mobility comparable to that of Au–DNA–Mb constructs (lane 2). Finally, additional controls were preformed to rule out non specific interaction of native Mb with AuNP–L1, AuNP–L1–DNA and AuNP–L1–D–heme and no change in electrophoretic mobility was observed upon addition to the protein.
To confirm that fully functional multi-AuNP hybrid is obtained using trifunctional linker, first, the hybridization ability of attached DNA was explored. After incubation of AuNP–L1–D with cDNA at room temperature for 4 h, the successful hybridization was demonstrated by a change in electrophoretic mobility and ethidium bromide staining (Fig. 4a). Subsequently, the peroxidase activity of attached Mb was investigated using oxidation of non fluorescent substrate Ampliflu Red into fluorescent resorufin (Fig. 4b) in the presence of H2O2.39 In such way, enzymatic activity of Mb can directly be assessed using fluorescence measurements.
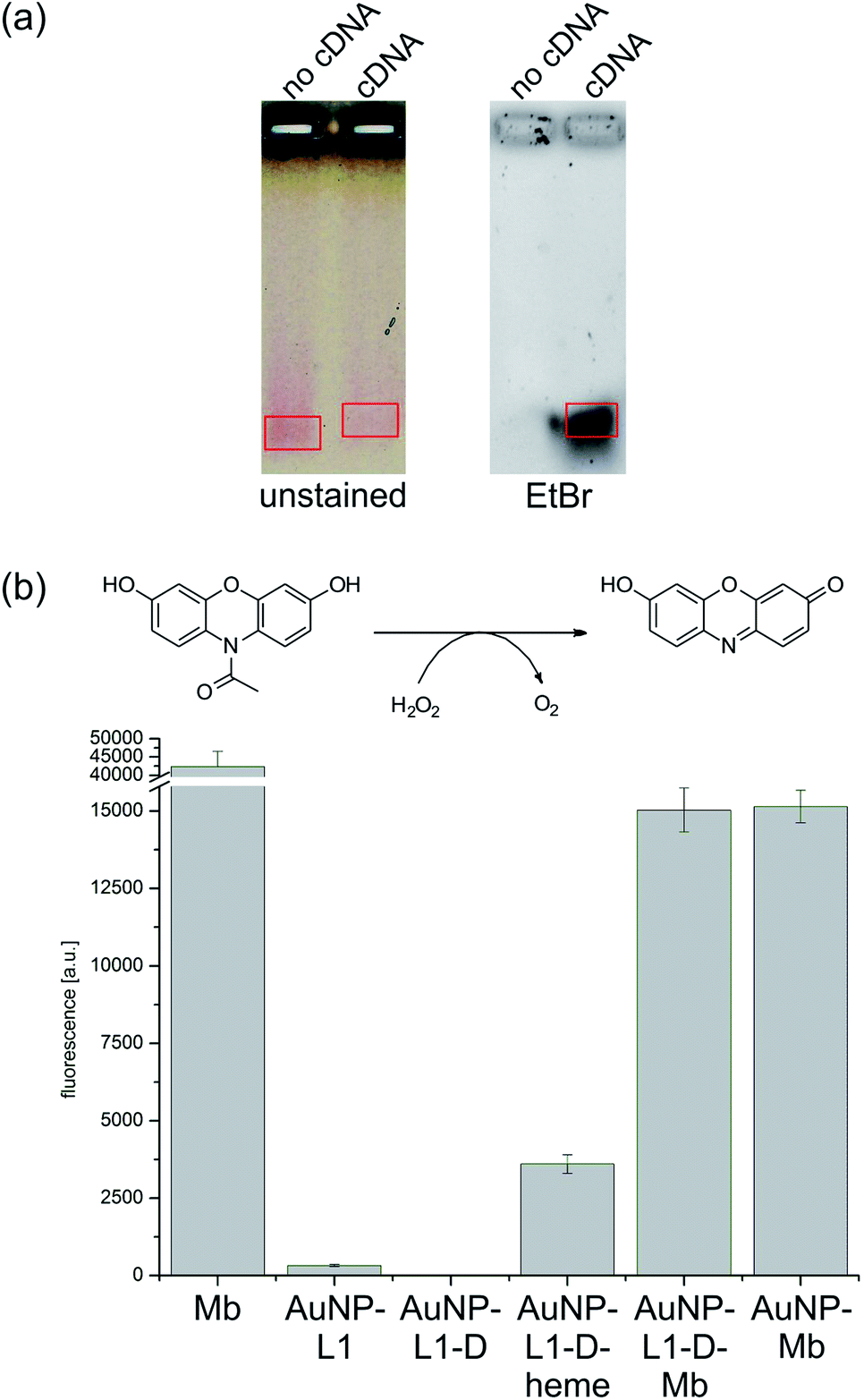 | ||
| Fig. 4 (a) 1% agarose gel of hybridization of AuNP–L1–D with cDNA before (left) and after ethidium bromide staining (EtBr, right). (b) Enzymatic activity by AmpliFlu Red assay. Shown are the measured fluorescence signals for different precursors towards the assembly of AuNP–DNA–Mb conjugates after a reaction time of 15 min. | ||
Measured peroxidase activity of AuNP–L1–D–heme after incubation with apoMb clearly demonstrates that a functional enzyme is present on the surface of the NP. In addition, the activity of the Mb reconstituted on the surface of AuNPs is comparable to that of the control, covalently bound AuNP–Mb conjugates. Furthermore, AuNP–L1, AuNP–L1–D and AuNP–L1–D–heme have negligible or low activity in comparison to AuNP–L1–D–Mb. Native Mb appears to be twice as active as both AuNP–Mb and AuNP–L1–D–Mb hybrids, which might be a consequence of the reduced measured fluorescence due to the quenching effects of the AuNP.
Conclusions
Herewith we describe the synthesis of multifunctional linkers for one-pot synthesis of stable AuNPs, which can further be modified with different molecular species. AuNP containing orthogonal functional groups were used to prepare AuNP conjugates containing both DNA and heme cofactor. Heme can be employed as an anchor for number of proteins through reconstitution as demonstrated by reconstitution of apo myoglobin. Using multifunctional linker, fully functional Au–DNA–Mb hybrids were obtained. Such multifunctional constructs can expand the NP toolbox for biosensing and assembly of novel catalytical materials as well as aide the preparation of novel multifunctional “target and deliver” therapeutic agents, which is the subject of our ongoing studies.Experimental
Materials and method
All chemicals were purchased from Sigma-Aldrich and used without further purification. Azido–DNA was obtained from Atdbio (Southampton, UK).Synthesis of linker
Multifunctional linkers were prepared starting from commercially available Fmoc-Lys(Boc)-OH (Scheme 1). Details on the synthesis of each component are presented in ESI.†Preparation of Au NPs
For the synthesis of AuNPs modified protocol of previously reported strategy was employed.22 Briefly, all glassware was cleaned thoroughly with aqua regia, washed with water and dried in an oven at 70 °C. In a typical synthesis 312 μL HAuCl4·H2O (50.8 mM, 15.8 μmol, 20 eq.) were stirred in 50 mL double distilled water with 39.6 μL linker (20 mM, 0.79 μmol, 1 eq.) for 1 h at room temperature. 144 μL NaBH4 (880 mM, 126.4 μmol, 160 eq.) were added in 4 steps over the course of 30 min. The solution was stirred over night and analyzed with UV-vis (Cary 300 Scan (Varian) scanned from 200–800 nm with water as reference).TEM study
The TEM samples were prepared by ultrasonic dispersion of the particle solution on a carbon support film (3 nm, TED Pella Inc. California). TEM experiments were performed using a Philips CM200FEG/ST microscope. Diameters were calculated from N = 100 with the help of ImageJ. Concentrations of AuNP were calculated based on the NP size using reported extinctions coefficients22 at the absorption maximum of 520 nm and were usually in the range of 100 nM.Coupling of DNA to AuNPs
The Cu-catalyzed Huisgen cycloaddition was performed using previously published approach.23 In a typical reaction 5 mL AuNP–L1 (100 μM, 0.5 nmol, 1 eq.) were incubated with 50 μL azDNA (100 μM, 5 nmol, 10 eq., sequence: (T)8 GAT CTC TTC ACC), 2.5 μL sodium ascorbate (40 mM, 100 mol, 200 eq.) and a premixed solution of 300 μL TBTA (333 μM in H2O–DMSO–tBuOH 4:3:1, 100 nmol, 200 eq.) and 2 μL copper sulfate pentahydrate (25 mM, 50 nmol, 100 eq.). The reaction mixture was incubated over night at 4 °C. To stabilize the DNA on the surface 2 mL TETBS300 (20 mM Tris–HCl, 300 mM NaCl, 5 mM EDTA, 0.05% Tween-20, pH 7.5) was added to the mixture and left to incubate for 2 h at room temperature before purification by several centrifugation and wash steps (20 min, 13200 rpm). The residue was suspended in TETBS300 to yield a red solution.
Heme coupling
Heme carboxy groups (5 μL heme, 5 mM, 25 nmol, 1000 eq.) were activated with 2.5 μL HBTU (15 mM, 37.5 nmol, 1500 eq.), 1.67 μL HOBt (30 mM, 50 nmol, 2000 eq.) and 4.5 μL DIPEA (20 mM, 90 nmol, 3600 eq.) for 15 min at room temperature. After addition of 200 μL AuNP–L1–D (125 nM, 0.025 nmol, 1 eq.) the reaction was left over night at room temperature and afterwards purified by several centrifugation cycles (20 min, 13200 rpm).
Reconstitution of myoglobin (Mb)
ApoMb was prepared by a reported procedure,24 namely by dissolving around 2.5 mg Mb from equine skeletal muscle Mb in 200 μL water and adjusting the pH to 2 by adding 0.1 M HCl. After addition of ice cold 2-butanone, the heme could be extracted from aqueous phase, aqueous batches were joined and buffer exchanged to phosphate buffer by gel filtration with a NAP 5 column (GE Healthcare). 28 μL of freshly prepared apoMb (19 μM, 530 pmol, 200 eq.) was reconstituted with 50 μL AuNP–L1–D–heme (53 nM, 2.65 pmol, 1 eq.).Hybridization of Au–L1–DNA–Mb with complementary DNA cDNA
To prove that the oligonucleotide was intact after attachment onto AuNP, hybridization experiments were carried out using a complementary ssDNA cDNA (5′-CTTCACGATTGCCACTTTCCAC-3′) added in a 100 fold excess in water and left to hybridize for 4 h. Gel electrophoresis showed a shift in electrophoretic mobility and the staining with ethidium bromide (EtBr) additionally proved the formation of dsDNA.Covalent coupling of Mb to AuNP
To activate the carboxyl groups 0.165 μL of EDC and NHS (each, 0.1 M, 16.5 nmol, 500 eq.) was added to 1 mL AuNP-01:10 (33 nM, 33 pmol, 1 eq.) for 15 min. Followed by coupling of 116 μL of Mb (285 μM, 33 nmol, 100 eq.). AuNP–Mb was purified by centrifugation (15 min, 13200 rpm) and re-suspended in phosphate buffer.
Mb activity assay
To test for the peroxidase-like activity of Mb, 50 μL of a solution containing 10 μM AmpliFluRed™ and 100 μM H2O2 in sodium phosphate buffer (pH 6) were added to 50 μL of each sample (0.3 μM, 15 pmol). Formation of resorufin was followed by measuring fluorescence emission at 590 nm after excitation at 530 nm.Acknowledgements
Presented work was supported by FET grant Single Molecule Activation and Computing (FOCUS), grant agreement no 270483 as well as DFG-CFN project A5.7 We would like to thank Pascal Bockstaller for his help with TEM measurements.Notes and references
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- X. Zheng, Q. Liu, C. Jing, Y. Li, D. Li, W. Luo, Y. Wen, Y. He, Q. Huang, Y. T. Long and C. Fan, Angew. Chem., 2011, 50, 11994–11998 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1333 CrossRef.
- A. E. Prigodich, A. H. Alhasan and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 2120–2123 CrossRef CAS PubMed.
- I. H. Lee, H. K. Kwon, S. An, D. Kim, S. Kim, M. K. Yu, J. H. Lee, T. S. Lee, S. H. Im and S. Jon, Angew. Chem., 2012, 51(35), 8800–8805 CrossRef CAS PubMed.
- V. Biju, Chem. Soc. Rev., 2014, 43, 744–764 RSC.
- H. Cheng, L. Yang, Y. Jiang, Y. Huang, Z. Sun, J. Zhang, T. Hu, Z. Pan, G. Pan, T. Yao, Q. Bian and S. Wei, Nanoscale, 2013, 5, 11795–11800 RSC.
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- K. Park, J. Jeong and B. H. Chung, Chem. Commun., 2012, 48, 10547–10549 RSC.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- A. M. Alkilany, L. B. Thompson, S. P. Boulos, P. N. Sisco and C. J. Murphy, Adv. Drug Delivery Rev., 2012, 64, 190–199 CrossRef CAS PubMed.
- J. Jeong, C. S. Lee, S. J. Chung and B. H. Chung, Bioprocess Biosyst. Eng., 2010, 33, 165–169 CrossRef CAS PubMed.
- Y. Ren, H. Zhang, B. Chen, J. Cheng, X. Cai, R. Liu, G. Xia, W. Wu, S. Wang, J. Ding, C. Gao, J. Wang, W. Bao, L. Wang, L. Tian, H. Song and X. Wang, Int. J. Nanomed., 2012, 7, 2261–2269 CAS.
- J. Conde, A. Ambrosone, V. Sanz, Y. Hernandez, V. Marchesano, F. Tian, H. Child, C. Berry, M. R. Ibarra Garcia, P. V. Baptista, C. Tortiglione and J. M. De La Fuente, ACS Nano, 2012, 6(9), 8316–8324 CrossRef CAS PubMed.
- R. P. Brinas, A. Sundgren, P. Sahoo, S. Morey, K. Rittenhouse-Olson, G. E. Wilding, W. Deng and J. J. Barchi Jr, Bioconjugate Chem., 2012, 23, 1513–1523 CrossRef CAS PubMed.
- V. Voliani, S. Luin, F. Ricci and F. Beltram, Nanoscale, 2010, 2, 2783–2789 RSC.
- P. A. G. Cormack, A. Hernandez-Santana, R. A. Prasath, F. McKenzie, D. Graham and W. E. Smith, Chem. Commun., 2008, 2517–2519 RSC.
- L. Fruk, C. H. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem., 2009, 48, 1550–1574 CrossRef CAS PubMed.
- E. Oh, K. Susumu, R. Goswami and H. Mattoussi, Langmuir, 2010, 26, 7604–7613 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- F. W. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef CAS.
- M. Miljevic, B. Geiseler, T. Bergfeldt, P. Bockstaller and L. Fruk, Adv. Funct. Mater., 2014, 24, 907–915 CrossRef CAS.
- M. D. Best, Biochemistry, 2009, 48, 6571–6584 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC.
- S. L. Khatwani, J. S. Kang, D. G. Mullen, M. A. Hast, L. S. Beese, M. D. Distefano and T. A. Taton, Bioorg. Med. Chem., 2012, 20, 4532–4539 CrossRef CAS PubMed.
- A. Heuer-Jungemann, R. Kirkwood, A. H. El-Sagheer, T. Brown and A. G. Kanaras, Nanoscale, 2013, 5, 7209–7212 RSC.
- P. N. Njoki, I. I. S. Lim, D. Mott, H. Y. Park, B. Khan, S. Mishra, R. Sujakumar, J. Luo and C. J. Zhong, J. Phys. Chem. C, 2007, 111, 14664–14669 CAS.
- D. Baranov and E. N. Kadnikova, J. Mater. Chem., 2011, 21, 6152–6157 RSC.
- Y.-W. Lin and J. Wang, J. Inorg. Biochem., 2013, 129, 162–171 CrossRef CAS PubMed.
- I. Matsunaga and Y. Shiro, Curr. Opin. Chem. Biol., 2004, 8, 127–132 CrossRef CAS PubMed.
- A. Onoda, Y. Kakikura, T. Uematsu, S. Kuwabata and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 2628–2631 CrossRef CAS.
- K. Oohora, Y. Kihira, E. Mizohata, T. Inoue and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 17282–17285 CrossRef CAS PubMed.
- J. Jiang, Y. He, X. Yu, J. Zhao and H. Cui, Anal. Chim. Acta, 2013, 791, 60–64 CrossRef CAS PubMed.
- E. Golub, R. Freeman, A. Niazov and I. Willner, Analyst, 2011, 136, 4397–4401 RSC.
- L. Fruk and C. M. Niemeyer, Angew. Chem., 2005, 44, 2603–2606 CrossRef CAS PubMed.
- L. Fruk, J. Muller, G. Weber, A. Narvaez, E. Dominguez and C. M. Niemeyer, Chem.–Eur. J., 2007, 13, 5223–5231 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of trifunctional linker and data for the optimization of nanoparticle synthesis. See DOI: 10.1039/c4ra01773k |
| This journal is © The Royal Society of Chemistry 2014 |