Facile synthesis of enol ethers via Zn(OTf)2-mediated formal alkyne hydration-Smiles rearrangement†
Xinying Chewa,
Yuhan Linb and
Yee Hwee Lim*a
aOrganic Chemistry, Institute of Chemical and Engineering Sciences (ICES), Agency for Science, Technology and Research (A*STAR), 11 Biopolis Way, #03-08 Helios Block, Singapore 138667, Singapore. E-mail: lim_yee_hwee@ices.a-star.edu.sg
bSingapore Bioimaging Consortium (SBIC), Agency for Science, Technology and Research (A*STAR), 11 Biopolis Way, #01-02 Helios Block, Singapore 138667, Singapore
First published on 26th March 2014
Abstract
An efficient protocol involving commercially available zinc trifluoromethanesulfonate was able to transform a terminal alkyne to an enol ether via a formal tandem Markovnikov hydration-Smiles type rearrangement. The chemoselective reaction was able to work with a range of heteroaromatic tethers such as diazine, pyridine, benzimidazole and benzothiazole.
Alkyne hydration or the addition of water to an alkyne is an atom-economical1 method to generate useful carbonyl compounds and therefore is a subject of much interest. Since the discovery of mercury(II) salts that hydrate alkynes with Markovnikov selectivity to give ketones,2 most of the research has focused on the development of less toxic, more selective and efficient metal and acid catalysts or biocatalysts for this process.3 Non-metal based catalysts such as formic acid,4 triflic acid5 or trifluoroacetic acid6 can be used to hydrolyze terminal alkynes to the Markovnikov ketone product at elevated temperatures in water. Numerous metal salts and complexes have also been identified to convert terminal alkynes to ketones (Markovnikov product) or aldehydes (anti-Markovnikov product), (Scheme 1) with the most prominent being palladium(II),7 gold(I),7e,8 gold(III),9 platinum(II)10 and ruthenium11 catalysts. Catalytic anti-Markovnikov alkyne hydration is a relatively new and exciting area where ruthenium12 has emerged to be a highly efficient catalyst.
 | ||
| Scheme 1 General scheme of terminal alkyne hydration. | ||
Being in the same group as mercury, studies using zinc for alkyne hydrations was reported as early as 1909 by Kutscheroff2c but was found to require very harsh conditions.13 There were few reported research using zinc thereafter. In 1994, an anti-Markovnikov hydration using silica gel-supported zinc borohydride to afford terminal alcohols rather than the aldehyde was disclosed.14
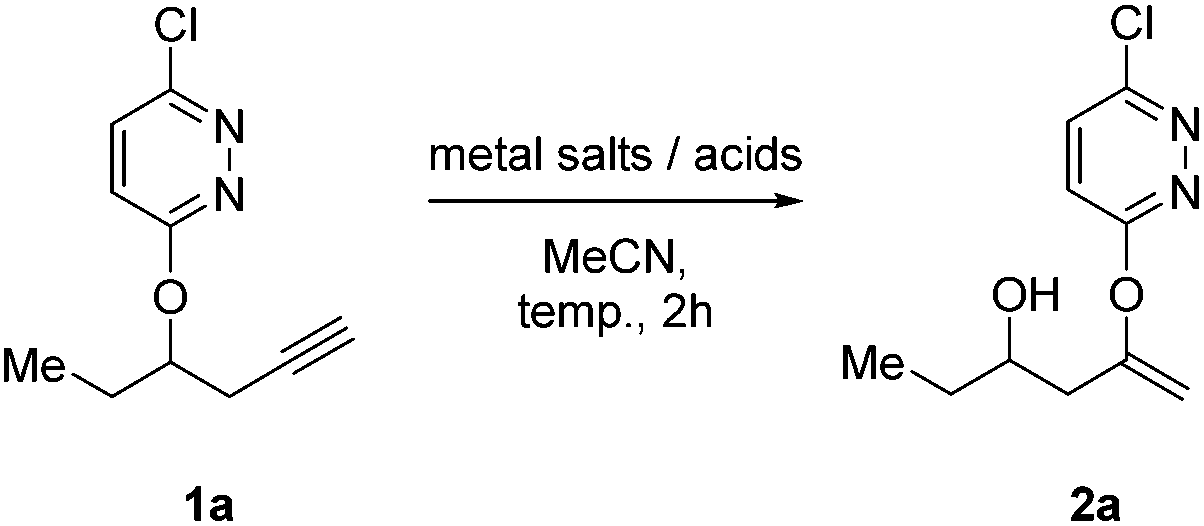
During the course of our investigations on diazines, we discovered an extremely facile protocol to convert the terminal alkynes to enol ethers using cheap and commercially available reagent, zinc triflate (Scheme 2). We believed that the terminal alkyne 1a was first hydrated in a Markovnikov manner to give an intermediate A that underwent an intramolecular Smiles-type rearrangement15 to yield the diazine-protected enol ether 2a. No ketone was ever observed or isolated.
 | ||
| Scheme 2 Zinc-mediated formal alkyne hydration-Smiles rearrangement of diazine 1a. | ||
An initial experiment was performed using 1a with Zn(OTf)2 in MeCN at ambient temperature. To our pleasant surprise, the reaction proceeded cleanly to give new product in nearly quantitative yield (95%) after 16 h. After extensive NMR, mass and IR spectroscopic analyses, the product was characterized to be 2a.16 As Zn(OTf)2 is not known to mediate alkyne hydration nor Smiles rearrangement to the best of our knowledge, we decided to further investigate this serendipitous observation.
A range of metal salts and acids commonly known to effect alkyne hydrations were first screened to determine if the observed reactivity behavior was exclusive (Table 1).17 It was found that a variety of other metal salts such as AgNTf2, AgOTf18 and CuOTf could yield the same product 2a (Table 1, entries 2, 3 and 9–11). A control experiment with triflic acid was carried out to determine whether the reaction was mediated by Brønsted acid and only a small amount (7%) of the desired product could be obtained with catalytic amount of TfOH (Table 1, entry 8). Zn(OTf)2 was by far superior in terms of efficiency and yield (Table 1, entries 1 and 12). At elevated temperature (80 °C), the reaction could be completed within 2 h to give 2a in 95% NMR yield (Table 1, entry 12). Stoichiometric amount of Zn(OTf)2 was necessary to effect the reaction as the conversion and yield dropped drastically with sub-stoichiometric amount of Zn(OTf)2 (Table 1, entry 13).
|
||||
|---|---|---|---|---|
| Entry | Metal salt/acid | Temp. (°C) | Conv.b (%) | Yieldb (%) |
| a Conditions: 1a (1 equiv.), metal salt/acid (1 equiv.), MeCN (0.1 M), 2 h; then H2O, 80 °C, 1 h.b Determined by 1H NMR with 2,4,6-collidine as an internal standard.c 5 mol% of TfOH was used instead.d 50 mol% of Zn(OTf)2 was used instead. | ||||
| 1 | Zn(OTf)2 | 25 | 60 | 50 |
| 2 | AgOTf | 25 | 15 | 10 |
| 3 | CuOTf | 25 | 45 | 25 |
| 4 | FeCl3 | 25 | 0 | 0 |
| 5 | PdCl2 | 25 | 100 | 0 |
| 6 | CuF2 | 25 | 10 | 0 |
| 7 | RuCl3 | 25 | 100 | 0 |
| 8c | TfOH | 80 | 16 | 7 |
| 9 | AgNTf2 | 80 | 80 | 40 |
| 10 | AgOTf | 80 | 50 | 45 |
| 11 | CuOTf | 80 | 100 | 40 |
| 12 | Zn(OTf)2 | 80 | 100 | 95 |
| 13d | Zn(OTf)2 | 80 | 48 | 45 |
Different polar solvents were screened to determine the effect of solvents on the reaction (Table 2). Whilst solvents like MeOH, acetone, 1,4-dioxane and EtOAc were found to give some conversion (Table 2, entries 2, 4, 5 and 6), MeCN was found to be the best solvent. It is interesting to note that the reaction proceeded even in pure water, albeit with very low conversion (Table 2, entry 7). We then studied the water tolerance level of this reaction (Table 2, entries 8–12). For reactions in a mixture of MeCN and water, the efficiency of the reaction decreases as the water content increases. At 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN–H2O mixture, the conversion dropped to 75% whilst the yield was only 55% (Table 2, entry 10).
:1 MeCN–H2O mixture, the conversion dropped to 75% whilst the yield was only 55% (Table 2, entry 10).
|
|||
|---|---|---|---|
| Entry | Solvent | Conv.b (%) | Yieldb (%) |
| a Conditions: 1a (1 equiv.), Zn(OTf)2 (1 equiv.), solvent (0.1 M), 80 °C, 2 h; then H2O, 80 °C, 1 h.b Determined by 1H NMR with 2,4,6-collidine as an internal standard. | |||
| 1 | MeCN | 100 | 95 |
| 2 | MeOH | 15 | 10 |
| 3 | DMSO | 0 | 0 |
| 4 | Acetone | 100 | 25 |
| 5 | 1,4-Dioxane | 100 | 20 |
| 6 | EtOAc | 100 | 40 |
| 7 | H2O | 10 | 5 |
| 8 | MeCN–H2O (9:1) |
100 | 75 |
| 9 | MeCN–H2O (4:1) |
90 | 70 |
| 10 | MeCN–H2O (1:1) |
75 | 55 |
| 11 | MeCN–H2O (1:4) |
0 | 0 |
| 12 | MeCN–H2O (1:9) |
0 | 0 |

In order to confirm that an external molecule of water was added during the hydration-rearrangement, 1c was treated with Zn(OTf)2 in MeCN at 80 °C for 2 h followed by a few drops of enriched 18O[H2O]. Indeed, mass spectroscopic analysis of the product 18O-2c found incorporation of heavy water, affording a product of m/z 225.0278 for [M + Na]+.19 Based on the above optimized conditions of using stoichiometric amount of Zn(OTf)2 in MeCN at 80 °C, the scope of the reaction was then examined.20
A range of diazines 1 were explored (Table 3). In general, the reactions proceeded smoothly with excellent yields (85–97%) to afford the corresponding enol ethers. The reaction was found to be tolerant of ether (Table 3, entry 4) as well as alkene functional groups (Table 3, entry 7). As expected, the stereochemistry of 1d was retained in the hydration-rearrangement process (Table 3, entry 4).21 Excellent chemoselectivity was observed as demonstrated by substrate 1f where the integrity of the second alkyne remained intact under the reaction conditions (Table 3, entry 6). The expected tertiary alcohol product for substrate 1g was not obtained; instead the dehydrated triene 2g was isolated (Table 3, entry 7).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Tb (h) | Yieldc (%) |
| a Conditions: substrate 1 (1 equiv.), Zn(OTf)2 (1 equiv.), MeCN (0.1 M), 80 °C, for specified time; then H2O (1 mL mmol−1), 80 °C for 1 h.b Reaction was monitored by TLC until all starting material was consumed.c Isolated yields.d The dehydrated product 2g was isolated instead of the expected tertiary alcohol. | ||||
| 1 |  |
 |
2 | 97 |
| 2 |  |
 |
2 | 95 |
| 3 |  |
 |
2 | 95 |
| 4 |  |
 |
3 | 88 |
| 5 |  |
 |
3 | 86 |
| 6 |  |
 |
3.5 | 85 |
| 7 | 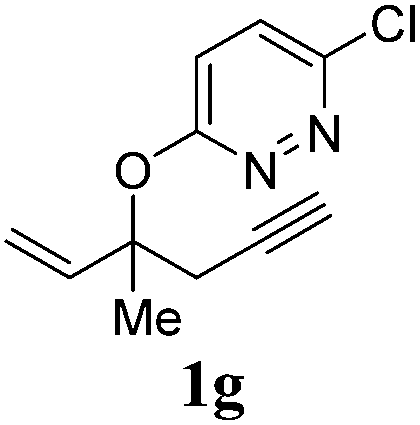 |
 |
3 | 87 |
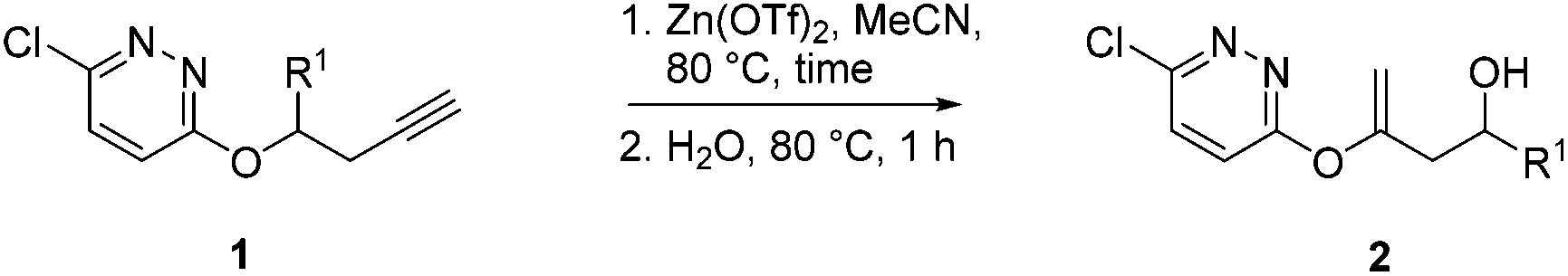
The presence of the diazine was vital for the success of the hydration-rearrangement reaction. Apart from diazine as the tether for the hydration-rearrangement, we decided to explore if other similar aromatic groups could be used. Numerous functionalities were attempted, Table 4 detailed some of the results.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Tb (h) | Yieldc (%) |
| a Conditions: substrate 3 (1 equiv.), Zn(OTf)2 (1 equiv.), MeCN (0.1 M), 80 °C, for specified time; then H2O (1 mL mmol−1), 80 °C for 1 h.b Reaction was monitored by TLC until all starting material was consumed.c Isolated yields.d 25% of side product 4c′ was obtained. | ||||
| 1 |  |
 |
24 | 89 |
| 2 |  |
 |
24 | 0 |
| 3 |  |
 |
3 | 45d |
| 4 |  |
 |
6 | 92 |
| 5 | 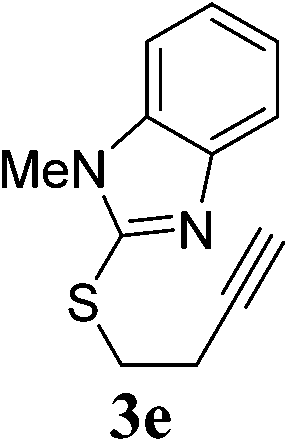 |
 |
6 | 81 |

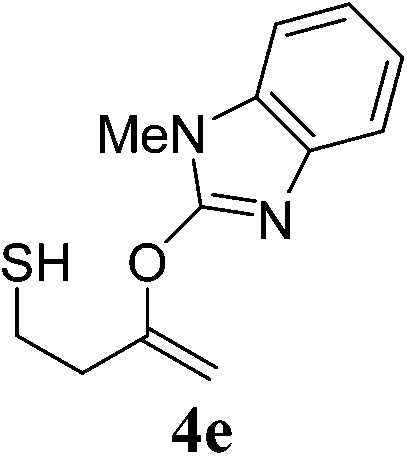
The pyridinyl 3a proceeded smoothly to afford the corresponding enol ether 4a in excellent yields (89%) (Table 4, entry 1). Benzoxazole ether 3b did not react while the corresponding benzoxazole thioether 3c gave the expected product 4c in 45% yield together with another by-product 4c′ in 25% (Table 4, entries 2 and 3). 4c′ was presumed to be formed through a transfer of the benzoxazole group from another starting molecule 3c. A similar reaction involving benzothiazole thioether 3d afforded the expected enol ether 4d in 92% yield in 6 h (Table 4, entry 4). Similarly, the benzimidazole thioether 3e proceeded smoothly to give 4e in 81% yield (Table 4, entry 5). No side products were observed in both cases. This difference in reactivity remains unclear at this point and is subject to further investigation.

Conclusions
In summary, a mild and efficient protocol to transform terminal alkynes to enol ethers via a formal tandem Markonikov hydration-Smiles rearrangement using cheap and commercially available zinc triflate has been developed. The reaction is highly chemoselective and works in a range of heteroaromatic such as diazine, pyridine, benzimidazole, benzoxazole and benzothiazole. The present study also highlights Zn(OTf)2 as an eco-friendly reagent where the transformation is 100% atom-economical. Further investigations into the mechanism as well as other applications in synthesis are currently in progress.Acknowledgements
We thank Ms Doris Tan (ICES) for high resolution mass spectrometric (HRMS) assistance. Financial support for this work was provided by A*STAR Joint Council Organisation (JCO), Singapore under the JCO Career Development Award (CDA) grant JCO12302EG013 (to Y.H.L.). Dr Romain Bejot was gratefully acknowledged for helping to obtain a sample of enriched 18O[H2O] from Singapore Radiopharmaceuticals Ltd for our investigations.Notes and references
- B. M. Trost, Science, 1991, 254, 1471 CAS.
- (a) M. Kutscheroff, Chem. Ber., 1881, 14, 1540 CrossRef PubMed; (b) M. Kutscheroff, Chem. Ber., 1884, 17, 13 CrossRef PubMed; (c) M. G. Kutscheroff, Chem. Ber., 1909, 42, 2759 CrossRef PubMed.
- L. Hintermann and A. Labonne, Synthesis, 2007, 1121 CrossRef CAS PubMed.
- (a) N. Menashe, D. Reshef and Y. Shvo, J. Org. Chem., 1991, 56, 2912 CrossRef CAS; (b) N. Menashe and Y. Shvo, J. Org. Chem., 1993, 58, 7434 CrossRef CAS.
- T. Tsuchimoto, T. Joya, E. Shirakawa and Y. Kawakami, Synlett, 2000, 1777 CAS.
- (a) F. Persico and B. D. Wuest, J. Org. Chem., 1993, 53, 95 CrossRef; (b) B. Meseguer, D. Alonso-Dìaz, N. Griebenow, T. Herget and H. Waldmann, Angew. Chem., Int. Ed., 1999, 38, 2902 CrossRef CAS; (c) S. Duclos, H. Stoeckli-Evans and T. R. Ward, Helv. Chim. Acta, 2001, 84, 3148 CrossRef CAS.
- (a) A. Avshu, R. D. O'Sullivan, A. W. Parkins, N. W. Alcock and R. M. Countryman, J. Chem. Soc., Dalton Trans., 1983, 1619 RSC; (b) K. Utimoto, Pure Appl. Chem., 1983, 55, 1845 CrossRef CAS; (c) K. Imi, K. Imai and K. Utimoto, Tetrahedron Lett., 1987, 28, 3127 CrossRef CAS; (d) Y. Kataoka, O. Matsumoto, M. Ohashi, T. Yamagata and K. Tani, Chem. Lett., 1994, 1283 CrossRef CAS; (e) B. Liu and J. K. De Brabander, Org. Lett., 2006, 8, 4907 CrossRef CAS PubMed.
- (a) E. Mizushima, K. Sato, T. Hayashi and M. Tanaka, Angew. Chem., Int. Ed., 2002, 41, 4563 CrossRef CAS; (b) P. Roembke, H. Schmidbaur, S. Cronje and H. Raubenheimer, J. Mol. Catal., 2004, 212, 35 CrossRef CAS PubMed; (c) E. Mizushima, D. Cui, D. C. D. Nath, T. Hayashi and M. Tanaka, Org. Synth., 2006, 83, 55 CrossRef CAS.
- (a) R. O. C. Norman, W. J. E. Parr and C. B. Thomas, J. Chem. Soc., Perkin Trans. 1, 1976, 1983 RSC; (b) Y. Fukuda and K. Utimoto, J. Org. Chem., 1991, 56, 3729 CrossRef CAS; (c) R. Casado, M. Contel, M. Laguna, P. Romero and S. Sanz, J. Am. Chem. Soc., 2003, 125, 11925 CrossRef CAS PubMed.
- (a) J. Chatt, R. G. Guy and L. A. Duncanson, J. Chem. Soc., 1961, 827 RSC; (b) W. Hiscox and P. W. Jennings, Organometallics, 1990, 9, 1997 CrossRef CAS; (c) J. W. Hartman, W. C. Hiscox and P. W. Jennings, J. Org. Chem., 1993, 58, 7613 CrossRef CAS.
- (a) J. Halpern, B. R. James and A. L. W. Kemp, J. Am. Chem. Soc., 1961, 83, 4097 CrossRef CAS; (b) J. Halpern, B. R. James and A. L. W. Kemp, J. Am. Chem. Soc., 1966, 88, 5142 CrossRef CAS; (c) Y. Sasaki and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 1986, 790 RSC; (d) M. M. Taqui Khan, S. B. Halligudi and S. Shukla, J. Mol. Catal., 1990, 58, 299 CrossRef.
- For selected references, see: (a) M. Tokunaga and Y. Wakatsuki, Angew. Chem., Int. Ed., 1998, 37, 2867 CrossRef CAS; (b) M. Tokunaga and Y. Wakatsuki, Jap. Patent 11319576 A2, 1999, Chem. Abstr., 1999, 131, 352828; (c) M. Tokunaga, T. Suzuki, N. Koga, T. Fukushima, A. Horiuchi and Y. Wakatsuki, J. Am. Chem. Soc., 2001, 123, 11917 CrossRef CAS PubMed; (d) D. B. Grotjahn, C. D. Incarvito and A. L. Rheingold, Angew. Chem., Int. Ed., 2001, 40, 3884 CrossRef CAS; (e) D. B. Grotjahn and D. A. Lev, J. Am. Chem. Soc., 2004, 126, 12232 CrossRef CAS PubMed; (f) F. Boeck, T. Kribber, L. Xiao and L. Hintermann, J. Am. Chem. Soc., 2011, 133, 8138 CrossRef CAS PubMed.
- M. Ban and F. Ida, US Pat., 2791614, 1957, Chem. Abstr., 1957, 51, 85804.
- B. C. Ranu, A. Sarkar, M. Saha and R. Chakraborty, Tetrahedron, 1994, 50, 6579–6584 CrossRef CAS.
- (a) L. A. Warren and S. Smiles, J. Chem. Soc., Abstr., 1930, 1327 RSC; (b) W. E. Truce, E. M. Kreider and W. W. Brand, Org. React., 1970, 18, 99 CAS.
- For more details on the structural assignment of 2a, please see ESI.†.
- For detailed screening results, please see ESI.†.
- R. Das and D. Chakraborty, Appl. Organomet. Chem., 2012, 26, 722 CrossRef CAS PubMed.
- 2c: C8H9N2O2ClNa has [M + Na]+ calcd: 223.0245, found: 223.0249. 18O-2c: C8H9N2O18OClNa has [M + Na]+ calcd: 225.0293, found: 225.0278.
- Representative procedure: To a solution of 1a (182 mg, 1 mmol) in MeCN (10 ml) was added Zn(OTf)2 (365 mg, 1 mmol) and the reaction mixture was stirred at 80 °C until consumption of 1a. Water (1 mL) was then added and the reaction was stirred for another 1 h. Solvent was removed and the crude was diluted with EtOAc and washed with saturated NaHCO3. The crude was purified by column chromatography to afford the desired product 2a as a colorless oil (95%).
- See ESI† for more details on determination of absolute configuration.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00159a |
| This journal is © The Royal Society of Chemistry 2014 |