DOI:
10.1039/C4RA01367K
(Paper)
RSC Adv., 2014,
4, 16769-16776
Novel catalyst by immobilizing a phosphotungstic acid on polymer brushes and its application in oxidative desulfurization
Received
17th February 2014
, Accepted 24th March 2014
First published on 27th March 2014
Abstract
In the study, an HPW–PDMAEMA–SiO2 (phosphotungstic acid (HPW); poly-N,N-dimethylaminoethyl methacrylate (PDMAEAM)) catalyst was successfully synthesized. The synthesized HPW–PDMAEMA–SiO2 catalyst was characterized via XRD, TEM, FT-IR, TGA, and ICP-AES. The results show that the HPW active species retained its Keggin structure after immobilizing into polymer brushes. At optimal reaction conditions, the oxidative desulfurization conversion of dibenzothiophene reached 100%, and there was no significant catalytic performance decrease after six recycles. The excellent recoverability of the catalyst was attributed to the decreased leaching of the HPW active species caused by the strong interaction between the negative [PW12O40]3− ions and positive ammonium ions in the PDMAEMA polymer brushes.
1. Introduction
The ultra-deep desulfurization of diesel fuel has attracted increasing attention because of the stringent regulations of sulfur levels in many countries. Oxidative desulfurization (ODS) is considered as one of the most promising methods for the ultra-deep desulfurization of diesel fuel. In the ODS process, sulfur compounds, such as benzothiophene (BT), dibenzothiophene (DBT), and 4,6-dimethyldibenzothiophene (4,6-DMDBT), are oxidized to their corresponding sulfones, which can be easily separated from the diesel by using an extractant. Thus, ultra-deep desulfurization of diesel fuel is achieved.
Many investigations attempted to find an effective catalyst for the ODS process. Studies have reported that titanium-containing mesoporous silica could function as a highly efficient catalyst for the ODS of refractory aromatic sulfur compounds to produce ultra-low sulfur diesel.1,2 Metal oxides, such as MgO, CaO, and CuO, exhibit excellent catalytic activity for the decomposition of sulfur-containing components in the ODS process.3–5 In recent years, heteropolyacids (HPAs) with the Keggin structure have been widely investigated because of their oxidizing capability in the two-phase system. Li et al.6 reported a simple ODS system with the presence of homogeneous phosphotungstic acid (HPW) catalyst, an oxidant, and ionic liquids ([Bmim]BF4) to achieve a deep desulfurization effect of the model oil. Trakarnpruk et al.7 reported polyoxometalate catalysts in the ODS of diesel fuel, with hydrogen peroxide/acetic acid as an oxidant. A high desulfurization rate of 98% was achieved at a mild reaction condition. Although these homogeneous HPA catalysts exhibit excellent catalytic performance for the ODS process, they are difficult to be separated from the diesel fuel, which is the main obstacle for their industrial application at present.8 Immobilizing HPA onto the surface of a solid support is necessary to obtain a heterogeneous catalyst, which can be easily separated from the diesel fuel via filtration. This process enhances the recycling ability of the HPA catalyst. Yan et al.9 synthesized a mesoporous HPW/TiO2 catalyst by incorporating HPW into mesoporous TiO2 via an evaporation-induced, and self-assembly method. This catalyst was applied in the ODS process of the model oil. The desulfurization rate of DBT reached 95.2%, and the catalytic activity loss of the obtained HPW/TiO2 catalyst was negligible after regenerating thrice. Li et al. adopted silica dioxide (SiO2),10 silica-pillared clay (SPC),11 and hexagonal mesoporous silicate (HMS)12 as solid supports of HPW. The obtained catalyst exhibited high catalytic activity with a minimal loss in the active species during the recycling ODS process.
In recent years, solid supports functionalized with polymer brushes have attracted substantial attention to improve the recoverability of homogeneous catalysts in different catalysis areas.13,14 In the present study, we attempted to synthesize poly-N,N-dimethylaminoethyl methacrylate (PDMAEMA) polymer brushes on the surface of an SiO2 support. A novel ODS catalyst of HPW–PDMAEMA–SiO2 was obtained by impregnating HPW on the PDMAEMA–SiO2 support. DMAEMA has aliphatic tertiary amino groups. PDMAEMA forms a typical stimuli-responsive polymer that exhibits a combined temperature and pH sensitivity after polymerization or cross-linking. PDMAEMA is known to be a muco-adhesive polymer, which indicates that it is cationic in acidified media or when quaternized with an alkylating agent.15,16 Considering the presence of ammonium ions in the molecular chain of PDMAEMA polymer brushes, an electrostatic force exists between the positive ammonium ions and negative [PW12O40]3− ions. Hence, [PW12O40]3− ions could be tightly immobilized onto the molecular chain of PDMAEMA polymer brushes. Based on the above hypothesis, the leaching of the HPW active species of HPW–PDMAEMA–SiO2 catalyst can decrease in the ODS process, and the recoverability of the HPW–PDMAEMA–SiO2 can be enhanced.
2. Experimental
2.1. Materials
Silica nanoparticles with a 50 ± 5 nm diameter were acquired from Aladdin and were used after drying in vacuum at 110 °C for 24 h to remove physically adsorbed materials and chemicals. 3-Aminopropyl triethoxysilane (APTES; Aladdin, 99%) and 2-bromo-2-methylpropionyl bromide (Aladdin, 99%) were used as receivers. N,N-Dimethylaminoethyl methacrylate (DMAEMA; Aladdin, 99%) was filtered through a basic aluminum oxide column before each polymerization experiment to remove inhibitors.
2.2. Synthesis of HPW–PDMAEMA–SiO2
The PDMAEMA–SiO2 support was prepared by following a process similar to the one found in literature.17,18 First, 1 g of SiO2 was mixed with 4 mL of APTES in 20 mL of anhydrous toluene. The mixture was then immersed in an oil bath at 368 K for 12 h in a nitrogen atmosphere. The sample was centrifuged, washed with methanol, dried in a vacuum for 12 h, and labeled as SiO2–NH2. SiO2–NH2 (0.5 g) was dispersed in 100 mL of anhydrous tetrahydrofuran (THF). About 8 mL of triethylamine and 4 mL of 2-bromo-isobutyl bromide were then added to the system. The mixture was immersed in an ice-water bath to maintain the temperature at 273 K for 1 h. The sample was centrifuged, washed with THF, dried at 313 K for 12 h, and labeled as SiO2–Br. About 0.5 g SiO2–Br was dispersed in 25 mL of methanol, and 0.148 mol of DMAEMA monomer, 0.5 g of CuCl, and 0.72 mL of 2,2-bipyridine were then added to the mixture. The solution was immersed in an oil bath at 308 K for 24 h. The mixture was centrifuged, washed with water and methanol, dried, and labeled as PDMAEMA–SiO2.
The HPW–PDMAEMA–SiO2 support catalyst was prepared using the following procedure: glacial acetic acid (0.66 mL) and PDMAEMA–SiO2 (1 g) were mixed together in 100 mL of ethanol. About 10 mL of 0.44 mmol HPW solution was added dropwise, and the mixture was stirred for 24 h at room temperature. After the reaction, the sample was centrifuged, washed with ethanol, dried at 313 K for 10 h, and labelled as HPW–PDMAEMA–SiO2. The whole synthesis route of the HPW–PDMAEMA–SiO2 catalyst is shown in Fig. 1. HPW–SiO2 was also prepared using a similar procedure that utilized SiO2 as a solid support.
 |
| | Fig. 1 Synthetic route of the HPW–PDMAEMA–SiO2 catalyst. | |
2.3. Characterization
Powder X-ray diffraction (XRD) data was collected from a Bruker advanced D8 X-ray diffractometer with Cu-Kα irradiation (λ = 1.5406 Å) as source at 40 kV and 40 mA. Transmission electron microscopy (TEM) experiments were performed using Tecnai F30 field emission TEM. Fourier transform infrared (FT-IR) spectra were obtained using a Nicolet AVATAR 360. Thermo-gravimetric analysis (TGA) was performed using a Netzsch STA-449F3 (Jupiter OR, Germany) analyzer in a nitrogen atmosphere. The heating rates were typically at 10 °C min−1, and the gas flow of nitrogen through the system was 20 mL min−1. Inductively coupled plasma atomic emission spectra (ICP-AES) were obtained using a thermo scientific TCAP 6000 SERICS ICP spectrometer.
2.4. Catalytic evaluation of catalyst
Model oils containing 54 ppm sulfur were prepared by dissolving DBT compounds in n-octane. The ODS of the model fuel was performed in a three-necked flask equipped with a total reflux system. Unless otherwise specified, catalytic oxidation was performed at 333 K, and 10 mL of the model oil with 100 mg of the catalyst, a specific amount of 30% aqueous H2O2 solution, and 10 mL of methanol as solvent were used. The reactor was fixed in an oil bath at different temperatures with stirring and reflux. The solid catalyst was recovered via centrifugation and washed with methanol for recycling. The clarified oil was then collected, and the sulfur content was analyzed using a microcoulometric detector (WK-2D). The analysis conditions were as follows: injection amount of oil, 1.3 μL; flow rate of N2, 260 mL min−1; flow rate of O2, 150 mL min−1; gasification temperature, 1023 K; burning temperature, 1123 K; and equilibrium temperature, 923 K. The desulfurization rate of ODS was determined using the following equation:| |
 | (1) |
where Sint is the initial concentration of the sulfur component in the model oil, and Sres is the residual concentration of the sulfur component after the ODS process as detected by the microcoulometric detector.
3. Results and discussion
3.1. Characterization of the catalyst
Fig. 2 shows the (A) small-angle and (B) wide-angle XRD patterns of the SiO2, PDMAEMA–SiO2, and HPW–PDMAEMA–SiO2 samples. In Fig. 2A, all three samples display (100) diffraction peaks at 2θ = 2.4°, which correspond to the mesoporous structure of SiO2. These results demonstrate that the samples of PDMAEMA–SiO2 and HPW–PDMAEMA–SiO2 retain a uniform mesoporous structure regardless of the grafting of PDMAEMA brushes and loading of HPW species. As shown in Fig. 2B, the samples of SiO2 and PDMAEMA–SiO2 have obvious diffraction peaks at 2θ = 22.5°, which originated from the amorphous nature of the pore wall of SiO2 support.12,19 For the HPW–PDMAEMA–SiO2 catalyst, a wide diffraction peak in the range of 15° to 40° was observed, thereby indicating that HPW was successfully loaded onto the support of PDMAEMA–SiO2 with high dispersion.
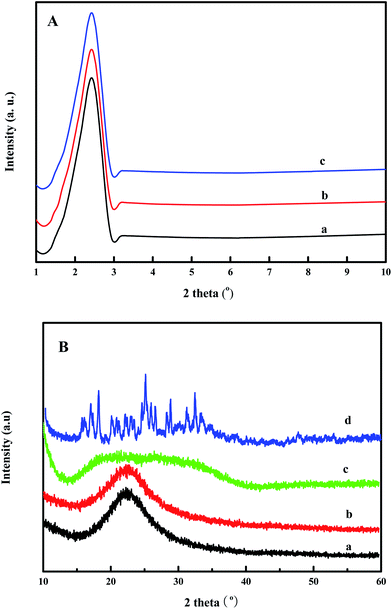 |
| | Fig. 2 (A) Small-angle and (B) wide-angle XRD patterns of (a) SiO2, (b) PDMAEMA–SiO2, (c) HPW–PDMAEMA–SiO2, and (d) HPW. | |
Fig. 3 shows the FT-IR spectra of SiO2, PDMAEMA–SiO2, HPW–PDMAEMA–SiO2, and HPW. The SiO2 sample displays framework bands at 805, 960, and 1060 cm−1, which correspond to the symmetric stretching frequency of Si–O–Si, stretching frequency of Si–O–H, and anti-symmetric stretching of Si–O–Si, respectively.10,12 After PDMAEMA was grafted on the surface of SiO2, the new strong absorption peak at 1730 cm−1 was ascribed to the stretching vibration of the ester carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in PDMAEMA brushes.19 HPW with the Keggin structure exhibited four typical IR bands at 1079, 983, 889, and 805 cm−1, which are assigned to the stretching frequencies of P–O, WO, W–Ob–W, and W–Oc–W bridges, respectively.11,12,20 For the HPW–PDMAEMA–SiO2 sample, the peaks at 1079, 983, and 805 cm−1 became stronger because of the overlap with the framework bands of SiO2. Moreover, it displayed a new peak at 889 cm−1. These results show that the HPW species retained its Keggin structure after loading onto the surface of PDMAEMA–SiO2.
O) in PDMAEMA brushes.19 HPW with the Keggin structure exhibited four typical IR bands at 1079, 983, 889, and 805 cm−1, which are assigned to the stretching frequencies of P–O, WO, W–Ob–W, and W–Oc–W bridges, respectively.11,12,20 For the HPW–PDMAEMA–SiO2 sample, the peaks at 1079, 983, and 805 cm−1 became stronger because of the overlap with the framework bands of SiO2. Moreover, it displayed a new peak at 889 cm−1. These results show that the HPW species retained its Keggin structure after loading onto the surface of PDMAEMA–SiO2.
 |
| | Fig. 3 FT-IR spectra of (a) SiO2, (b) PDMAEMA–SiO2, (c) HPW–PDMAEMA–SiO2, and (d) HPW. | |
The TEM images of SiO2, PDMAEMA–SiO2, and HPW–PDMAEMA–SiO2 samples are shown in Fig. 4. The particle size of SiO2 is approximately 20 nm and that of PDMAEMA–SiO2 is approximately 25 nm. The increasing particle size and the dark color on the surface of PDMAEMA–SiO2 sample indicate the presence of PDMAEMA brushes on SiO2. However, no obvious HPW species were observed in the images of HPW–PDMAEMA–SiO2 samples possibly because the HPW species is absorbed onto the surface in the form of [PW12O40]3− anion instead of a cluster. Thus, the said species is barely visible in Fig. 4c.
 |
| | Fig. 4 TEM images of (a) SiO2, (b) PDMAEMA–SiO2, and (c) HPW–PDMAEMA–SiO2. | |
Fig. 5 shows the TGA curves of the SiO2, HPW, PDMAEMA–SiO2, and HPW–PDMAEMA–SiO2 samples within the region of 303 K to 1073 K in N2 atmosphere. All samples were purified and dried before testing. The SiO2 sample exhibited a 2.5% weight loss when the temperature was increased to 873 K, which was attributed to the condensation of silanol groups to form siloxane bonds. The weight loss of PDMAEMA–SiO2 was approximately 9.4% from 523 K to 873 K. A 1.5% difference of the weight loss between the PDMAEMA–SiO2 and HPW–PDMAEMA–SiO2 samples was observed at 873 K. For the pure HPW species, the weight loss at 873 K was 4.5% because of water molecule loss and decomposition of HPW with the Keggin structure.21,22 Based on the above results, the calculated HPW content on the surface of the PDMAEMA–SiO2 support was approximately 33 wt%.
 |
| | Fig. 5 TGA analysis of (a) SiO2, (b) pure HPW, (c) PDMAEMA–SiO2, and (d) HPW–PDMAEMA–SiO2. | |
3.2. Catalytic performance of the catalyst
The ODS process was performed on a model fuel by using DBT dissolved in n-octane as the model sulfur compound and methanol as the extraction solvent to evaluate the effect of the synthesized HPW–PDMAEMA–SiO2 catalyst on desulfurization. Two blank runs were performed in absence of H2O2 oxidant and catalyst to demonstrate the influence of methanol extraction. The results are shown in Fig. 6, which shows that the desulfurization rate of the ODS system was approximately 26% with the extraction of methanol, and it reached 46% after H2O2 oxidant was added to the ODS system. The desulfurization rate of DBT significantly increased after adding the HPW–PDMAEMA–SiO2 catalyst to the ODS system. When the reaction time was 3.0 h, the desulfurization conversion rates of the HPW–PDMAEMA–SiO2 catalyst were almost 100%. These results illustrate the contribution of the HPW–PDMAEMA–SiO2 catalyst to the ODS system. The excellent catalytic activity of the HPW–PDMAEMA–SiO2 catalyst can be attributed to the presence of active HPW component. In the presence of excess H2O2, W(O)n complex in HPW was oxidized and disaggregated to form a peroxometal complex as W(O2)n, which provided catalytic activity for the ODS of the sulfur component. Therefore, the high desulfurization rate of DBT was mainly caused by the catalytic effect of the HPW species in the HPW–PDMAEMA–SiO2 catalyst and not by the extraction effect of methanol.
 |
| | Fig. 6 Effect of the catalyst on desulfurization rate. (a) Model oil + methanol, (b) model oil + methanol + H2O2, and (c) model oil + methanol + H2O2 + HPW–PDMAEMA–SiO2 catalyst. | |
The reaction conditions, including reaction time, catalyst dosage, O/S molecular ratio, and reaction temperature, were investigated to obtain the optimal desulfurization effect of DBT. As shown in Fig. 7A, the desulfurization rate of DBT increased along with the reaction time, and DBT was completely oxidized to its sulfones when the reaction time reached 2.5 h. The dosage of the HPW–PDMAEMA–SiO2 catalyst in the mixture of the 10 mL model fuel and 10 mL methanol was also investigated. Fig. 7B displays that the desulfurization rate was enhanced along with catalyst dosage. In addition, the curve saturates when the catalyst dosage was 0.1 g per 10 mL. This result demonstrates that a catalyst concentration of 0.1 g per 10 mL in the model oil can provide enough active sites for the ODS process of DBT. As shown in Fig. 7C, the O/S molar ratio has a strong influence on the DBT desulfurization rate. The desulfurization rate of DBT increased with O/S molar ratio up until O/S = 12 and then slightly decreased beyond this value. The stoichiometric O/S of DBT oxidation was 2, which means that 2 mol of hydrogen peroxide was consumed for 1 mol of the DBT compound, and DBT was oxidized to its corresponding sulfones.23 The optimal value of the O/S molar ratio was much higher than the stoichiometric O/S molar ratio. The excess consumption of hydrogen peroxide may be attributed to the mass transfer resistance in the presence of the long molecular chain of PDMAEMA brushes in the HPW–PDMAEMA–SiO2 catalyst. Therefore, the oxidant concentration should be high enough to obtain the optimal desulfurization effect. Fig. 7D exhibits the effect of temperature on the desulfurization rate of the HPW–PDMAEMA–SiO2 catalyst. The temperature increment can significantly promote the desulfurization rate in ODS for the HPW–PDMAEMA–SiO2 catalyst. When the temperature was increased from 303 K to 333 K, the desulfurization rate remarkably improved from 95.2% to 100%. The quantity of the formed peroxometal complex as W(O2)n increased with temperature, and its oxidative ability toward DBT was enhanced.23,24 A substantially high temperature causes the decomposition of H2O2,25,26 thereby decreasing the concentration of the W(O2)n complex in the HPW active species. This condition may explain the reduction in the desulfurization rate when the reaction temperature is above 333 K.
 |
| | Fig. 7 Effect of (A) reaction time, (B) catalyst dosage, (C) O/S molar ratio, and (D) reaction temperature on the desulfurization rate of DBT. T = 333 K, O/S = 12, catalyst dosage = 0.1 g/10 mL, t = 3 h. | |
Kinetic experiments were performed for the desulfurization of DBT at different temperatures. The agitation speed was fixed at 1000 rpm to minimize external mass transfer resistance. In the presence of excess of H2O2, the oxidation of sulfur compounds follows pseudo-first-order reaction kinetics, as shown in eqn (2).27,28
| |
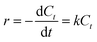 | (2) |
| |
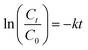 | (3) |
| |
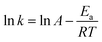 | (4) |
where
k is the pseudo-first-order reaction rate constant. Its value can be determined using the slope of the linear plot of ln(
C0/
Ct)
versus reaction time
t (
eqn (2)). A linear relationship was obtained in
Fig. 8A when ln(
C0/
Ct) was plotted
versus reaction time for temperatures of 303 K, 313 K, 323 K, and 333 K. From the reaction rate constant
k determined at various temperatures, the apparent activation energies for the oxidation of the DBT were calculated using the Arrhenius equation (
eqn (3)). As shown in
Fig. 8B, the apparent activation energy obtained for the desulfurization reaction is 23.2 kJ mol
−1. The activation energy of the HPW–PDMAEMA–SiO
2 catalyst was lower than the results reported in literature,
29 thereby indicating the excellent catalytic activity of the HPW–PDMAEMA–SiO
2 catalyst.
 |
| | Fig. 8 (A) Fitting of experimental data into the pseudo-first-order rate model. (B) Arrhenius activation energies for DBT of the HPW–PDMAEMA–SiO2 catalyst. | |
The recoverability of the heterogeneous catalyst is very important for its industrial application. The HPW–PDMAEMA–SiO2 catalyst was recycled six times to investigate its recovery rate. At the end of the reaction, the catalyst was recovered via filtration, washed with methanol several times, dried at 353 K, and subjected to the next ODS process. The results are shown in Fig. 9A. The desulfurization rate decreased from 100% to 97.8% after six recycles. The HPW–SiO2 catalyst, which was prepared by directly impregnating the HPW species on the SiO2 support, was also subjected to the ODS process for comparison. The desulfurization rate decreased from 99.2% to 75.9% after six recycles. These results indicate that the presence of PDMAEMA brushes in the HPW–PDMAEMA–SiO2 catalyst effectively enhanced its recoverability in the ODS process. The HPW species provides the primary active sites for the oxidation of DBT compounds to its corresponding sulfones. Therefore, the poor recoverability of the HPW–SiO2 catalyst may be attributed to the leaching of the HPW active species in the ODS process. In the synthesized HPW–PDMAEMA–SiO2 catalyst, the [PW12O40]3− anion can be tightly immobilized onto the molecular chain of PDMAEMA brushes because of the electronic force between the negative [PW12O40]3− ions and positive ammonium positive ions. Thus, the leaching of the HPW active species can be decreased, and the recoverability of the HPW–PDMAEMA–SiO2 is enhanced. Hot filtration experiments were performed in the ODS process to confirm that the leaching amount of HPW species from the HPW–PDMAEMA–SiO2 catalyst is low. The catalyst was filtered off when the reaction time reached 0.5 h, and its possible catalytic activity in the obtained solution at the same conditions was observed. Fig. 9B shows the catalytic activity for ODS after the hot filtration experiment compared with those in the presence of HPW–PDMAEMA–SiO2 and with the blank experiment without catalyst. After hot filtration of the catalyst, the desulfurization rate of the reaction system increased from 74.7% to 81.5% with reaction times between 0.5 h and 3 h, which were much lower than that with the HPW–PDMAEMA–SiO2 catalyst. This result indicates that only a small amount of the HPW active species dissolved from the PDMAEMA–SiO2 support in the ODS process.
 |
| | Fig. 9 (A) Effect of recycles on the desulfurization rate of (a) HPW–PDMAEMA–SiO2 and (b) HPW–SiO2 catalysts. (B) Hot filtration experiment of the HPW–PDMAEMA–SiO2 catalyst (a) without the presence of the HPW–PDMAEMA–SiO2 catalyst, (b) hot filtration of HPW–PDMAEMA–SiO2 at 0.5 h, and (c) with the presence of the HPW–PDMAEMA–SiO2 catalyst. Reaction conditions: temperature = 333 K; O/S = 8; catalyst amount = 0.06 g/10 mL; rpm = 600 rpm. | |
The HPW–PDMAEMA–SiO2 catalyst that was used for six cycles was characterized via XRD, and the results were compared with those of a fresh catalyst. From Fig. 10, the wide diffraction peak in the range of 15° to 40° could still be observed in the reused HPW–PDMAEMA–SiO2 catalyst, thereby indicating that the leaching of active HPW was low in the ODS process. The ICP-AES experiment was performed to measure the HPW content in the reused HPW–PDMAEMA–SiO2 and HPW–SiO2 catalysts after six cycles. The results are shown in Table 1, wherein the HPW content was 32.6 wt% in the fresh HPW–PDMAEMA–SiO2 catalyst, which is consistent with the results of TGA. The atomic ratio of P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W (the atomic ratio of phosphorus and tungsten elements) was 1:11.9, which is very close to the stoichiometric ratio of 1:12. The HPW content decreased from 32.6 wt% to 30.1 wt% with an approximately 2.5% loss in the HPW species with increasing recycling times of the catalyst, and the atomic ratio of P:W was maintained in the range of 11.9 to 10.5. This result demonstrates that the leaching of the HPW species of the HPW–PDMAEMA–SiO2 catalyst was rather low in the ODS process and was consistent with the result obtained from the hot filtration experiment. The low leaching of the HPW active species corresponds to the excellent recoverability of the HPW–PDMAEMA–SiO2 catalyst. The HPW content was 32.5 wt% in the fresh HPW–SiO2 catalyst. The HPW content decreased from 32.5 wt% to 23.3 wt% with approximately 9.2 wt% loss in HPW species with increasing recycling times of the catalyst, and the atomic ratio of P:W was maintained in the range of 11.8 to 10.5. This result demonstrates that the leaching of the HPW species of the HPW–SiO2 catalyst was rather high in the ODS process and was consistent with the result of the recycles on the desulfurization rate (Fig. 9A). The high leaching rate of the HPW active species corresponds to the poor recoverability of the HPW–SiO2 catalyst. These results show that the leaching of the HPW active site of HPW–PDMAEMA–SiO2 is much lower than that of the HPW–SiO2 catalyst, which is consistent with the excellent stability of HPW–PDMAEMA–SiO2 in the ODS process.
:W (the atomic ratio of phosphorus and tungsten elements) was 1:11.9, which is very close to the stoichiometric ratio of 1:12. The HPW content decreased from 32.6 wt% to 30.1 wt% with an approximately 2.5% loss in the HPW species with increasing recycling times of the catalyst, and the atomic ratio of P:W was maintained in the range of 11.9 to 10.5. This result demonstrates that the leaching of the HPW species of the HPW–PDMAEMA–SiO2 catalyst was rather low in the ODS process and was consistent with the result obtained from the hot filtration experiment. The low leaching of the HPW active species corresponds to the excellent recoverability of the HPW–PDMAEMA–SiO2 catalyst. The HPW content was 32.5 wt% in the fresh HPW–SiO2 catalyst. The HPW content decreased from 32.5 wt% to 23.3 wt% with approximately 9.2 wt% loss in HPW species with increasing recycling times of the catalyst, and the atomic ratio of P:W was maintained in the range of 11.8 to 10.5. This result demonstrates that the leaching of the HPW species of the HPW–SiO2 catalyst was rather high in the ODS process and was consistent with the result of the recycles on the desulfurization rate (Fig. 9A). The high leaching rate of the HPW active species corresponds to the poor recoverability of the HPW–SiO2 catalyst. These results show that the leaching of the HPW active site of HPW–PDMAEMA–SiO2 is much lower than that of the HPW–SiO2 catalyst, which is consistent with the excellent stability of HPW–PDMAEMA–SiO2 in the ODS process.
 |
| | Fig. 10 XRD patterns of (a) fresh PDMAEMA–SiO2 and (b) reused HPW–PDMAEMA–SiO2 catalysts. | |
Table 1 HPW content of HPW–SiO2 and HPW–PDMAEMA–SiO2 catalysts after different recycles
| Run number |
HPW–SiO2 |
HPW–PDMAEMA–SiO2 |
| P:Wa |
HPWb (wt%) |
P:Wa |
HPWb (wt%) |
| As determined by ICP-AES experiments. As calculated from the ICP-AES results, the atomic weight of P and W, and the molecular weight of HPW. |
| Fresh catalyst |
1:11.7 |
32.5 |
1:11.9 |
32.6 |
| 1 cycle |
1:11.7 |
31.2 |
1:11.7 |
32.5 |
| 2 cycle |
1:11.6 |
28.3 |
1:11.5 |
32.1 |
| 3 cycle |
1:11.8 |
25.2 |
1:11.8 |
31.4 |
| 4 cycle |
1:11.1 |
24.6 |
1:11.3 |
31.0 |
| 5 cycle |
1:10.9 |
23.9 |
1:11.8 |
30.7 |
| 6 cycle |
1:10.5 |
23.3 |
1:10.5 |
30.1 |
4. Conclusions
In this study, HPW–PDMAEMA–SiO2 catalyst was synthesized by functionalizing PDMAEMA polymer brushes on an SiO2 support. The presence of PDMAEMA polymer brushes tightly immobilized the HPW active species onto the surface of the support. The obtained HPW–PDMAEMA–SiO2 catalyst was applied in the ODS process of DBT. At optimal reaction conditions, the ODS conversion of DBT reached 100%, and no significant decrease in its catalytic performance was observed after six recycles. These results indicate that the HPW–PDMAEMA–SiO2 material is a promising and efficient heterogeneous catalyst for the ODS of diesel fuel.
Acknowledgements
The work was supported by the National Basic Research Program of China (973 Program, 2012CB720302), the Developing Program of Changjiang Scholar and Innovation Team from Ministry of Education of China (IRT1161), and Innovation Funds for Distinguished Young Scientists of Xinjiang Bingtuan (2011CD001).
References
- T. W. Kim, M. J. Kim, F. Kleitz, M. M. Nair, R. G. Nicolas, K. E. Jeong, H. J. Chae, C. U. Kim and S. Y. Jeong, ChemCatChem, 2012, 4, 687–697 CrossRef CAS PubMed.
- K. S. Cho and Y. K. Lee, Appl. Catal., B, 2014, 147, 35–42 CrossRef CAS PubMed.
- R. Sundararaman and C. S. Song, Appl. Catal., B, 2014, 148–149, 80–90 CrossRef CAS PubMed.
- R. Sundararaman and C. S. Song, Energy Fuels, 2013, 27, 6372–6376 CrossRef CAS.
- R. Sundararaman, X. L. Ma and C. S. Song, Ind. Eng. Chem. Res., 2010, 49, 5561–5568 CrossRef CAS.
- H. M. Li, L. N. He, J. D. Lu, W. S. Zhu, X. Jiang, Y. Wang and Y. S. Yan, Energy Fuels, 2009, 23, 1354–1357 CrossRef CAS.
- W. Trakarnpruk and K. Rujiraworawut, Fuel Process. Technol., 2009, 90, 411–414 CrossRef CAS PubMed.
- V. M. Z. Brenda, G. G. Álvaro, M. Poisot and R. G. Guillermo, Top. Catal., 2011, 54, 527–534 CrossRef.
- X. M. Yan, P. Mei, J. H. Lei, Y. Z. Mia, L. Xiong and L. P. Guo, J. Mol. Catal. A: Chem., 2009, 304, 52–57 CrossRef CAS PubMed.
- Z. E. A. Abdalla, B. S. Li and A. Tufail, Colloids Surf., A, 2009, 341, 86–92 CrossRef PubMed.
- B. S. Li, Z. X. Liu, C. Y. Han, J. J. Liu, S. L. Zuo, Z. Y. Zhou and X. M. Pang, J. Mol. Catal. A: Chem., 2011, 348, 106–113 CrossRef CAS PubMed.
- B. S. Li, W. Ma, J. J. Liu, C. Y. Han, S. L. Zuo and X. F. Li, Catal. Commun., 2011, 13, 101–105 CrossRef CAS PubMed.
- C. S. Gill, K. Venkatasubbaiah, N. T. S. Phan, M. Weck and C. W. Jones, Chem.–Eur. J., 2008, 14, 7306–7313 CrossRef CAS PubMed.
- L. Wei and C. W. Jones, ACS Catal., 2011, 1, 674–681 CrossRef.
- J. Niskanen, M. Karesoja, T. Rossic and H. Tenhu, Polym. Chem., 2011, 2, 2027–2036 RSC.
- Y. T. Joo, K. H. Jung, M. J. Kim and Y. Kim, J. Appl. Polym. Sci., 2013, 127, 1508–1518 CrossRef CAS PubMed.
- L. L. Zhou, W. Z. Yuan, J. Y. Yuan and X. Y. Hong, Mater. Lett., 2008, 62, 1372–1375 CrossRef CAS PubMed.
- J. Niskanen, C. Wu, M. Ostrowski, G. G. Fuller, S. Hietala and H. Tenhu, Macromolecules, 2013, 46, 2331–2340 CrossRef CAS.
- J. T. Sun, C. Y. Hong and C. Y. Pan, J. Phys. Chem. C, 2010, 114, 12481–12486 CAS.
- H. X. Zhang, J. J. Gao, H. Meng and C. X. Li, Ind. Eng. Chem. Res., 2012, 51, 6658–6665 CrossRef CAS.
- A. Llanos, L. Melo, F. Avendaño, A. Montes and J. L. Brito, Catal. Today, 2008, 133–135, 20–27 CrossRef CAS PubMed.
- S. Ajaikumar and A. Pandurangan, J. Mol. Catal. A: Chem., 2008, 286, 21–30 CrossRef CAS PubMed.
- D. S. Zhao, Y. N. Wang, E. H. Duan and J. Zhang, Fuel Process.
Technol., 2010, 91, 1803–1806 CrossRef CAS PubMed.
- W. S. Zhu, G. P. Zhu, H. M. Li, Y. H. Chao, Y. H. Chang, G. Y. Chen and C. R. Han, J. Mol. Catal. A: Chem., 2011, 347, 8–14 CrossRef CAS PubMed.
- Y. K. Park, S. Y. Kim, H. J. Kim, K. Y. Jung, K. E. Jeong, S. Y. Jeong and J. K. Jeon, Korean J. Chem. Eng., 2010, 27, 459–464 CrossRef CAS.
- D. Liu, J. Z. Gui, Y. K. Park, S. Yang, Y. H. Gao, X. L. Peng and Z. L. Sun, Korean J. Chem. Eng., 2012, 29, 49–53 CrossRef CAS PubMed.
- A. Sengupta, P. D. Kamble, J. K. Basu and S. Sengupta, Ind. Eng. Chem. Res., 2011, 51, 147–157 CrossRef.
- J. H. Ge, Y. M. Zhou, Y. Yang and M. W. Xue, Ind. Eng. Chem. Res., 2011, 50, 13686–13692 CrossRef CAS.
- J. H. Qiu, G. H. Wang, D. L. Zeng, Y. Tang, M. Wang and Y. J. Li, Fuel Process. Technol., 2009, 90, 1538–1542 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 












