
Theoretical investigation on the atmospheric fate of the CF3C(O)OCH(O)CF3 radical: alpha-ester rearrangement vs. oxidation†
Abstract
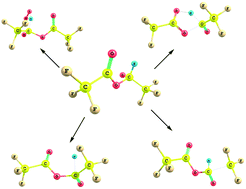
A detailed quantum chemical study is performed on the unimolecular decomposition reaction of the alkoxy radical, CF3C(O)OCH(O)CF3 produced from CF3C(O)OCH2CF3, trifluoroethyl trofluoroacetate (TFETFA) at the MPWB1K and M06-2X level of theories using the 6-31+G(d,p) basis set. Five plausible decomposition pathways including reaction with O2, α-ester rearrangement and thermal decomposition (C–C, C–H and C–O bonds scission) have been considered in detail. Out of the five prominent decomposition channels, our results reveal that reaction with O2 is the dominant path for the decomposition of the CF3C(O)OCH(O)CF3 radical in the atmosphere involving the lowest energy barrier which is in accordance with recent experimental findings. Our theoretical results also suggest that α-ester rearrangement leading to the formation of trifluoroacetic acid (TFA) has a negligible contribution for decomposition of the title alkoxy radical. The thermal rate constants for the above decomposition pathways are evaluated using Canonical Transition State Theory (CTST) at 298 K.
 Please wait while we load your content...
Please wait while we load your content...

