
How to easily adapt cloud points of statistical thermosensitive polyacrylamide-based copolymers knowing reactivity ratios†
Abstract
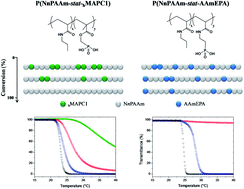
The present contribution deals with the ease of controlling the thermosensitivity of statistical copolymers prepared by free radical polymerization playing on the reactivity ratios of both monomers used for the synthesis. The copolymers were prepared using N-n-propylacrylamide (NnPAAm) monomer to achieve the thermoresponsive properties. The second monomer was either (dimethoxyphosphoryl)methyl 2-methylacrylate (MAPC1) or diethyl 2-(acrylamido)ethylphosphonate (DAAmEP) in order to incorporate phosphonic acid moieties after hydrolysis. The architecture of these original statistical copolymers was determined by measuring the reactivity ratios of both (NnPAAm/MAPC1) and (NnPAAm/DAAmEP) monomer couples. By varying the chemical nature of the phosphonated-based monomer (methacrylate or acrylamide), it was possible to get different reactivity ratios, and as a consequence to significantly affect the thermosensitive behavior of the statistical copolymers for a constant proportion of phosphonic acid groups. Additionally, all copolymers showed similar sorption rates toward nickel cations (Ni2+).
 Please wait while we load your content...
Please wait while we load your content...

