 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increased elasticity of a low-bandgap conjugated copolymer by random segmentation for mechanically robust solar cells†
Adam D.
Printz‡
,
Suchol
Savagatrup‡
,
Daniel J.
Burke
,
Trevor N.
Purdy
and
Darren J.
Lipomi
*
Department of NanoEngineering, University of California, San Diego, 9500 Gilman Drive, Mail Code 0448, La Jolla, CA 92093-0448, USA. E-mail: dlipomi@ucsd.edu
First published on 5th March 2014
Abstract
Despite the necessity of organic electronic materials to undergo large deformations in flexible, ultra-thin, and stretchable applications, many high-performance organic semiconductors are mechanically fragile. This paper describes an approach to increase the elasticity of low-bandgap conjugated polymers by statistical incorporation of unlike monomers. The material under study is PDPP2FT, an alternating copolymer. Synthesized by the Stille polymerization, it comprises an N-alkylated diketopyrrolopyrrole (DPP) unit flanked by two furan rings (2F) alternating with thiophene (T). In the modified (“segmented”) polymer, PDPP2FT-seg-2T, the DPP is exchanged for a tail-to-tail coupled unit of two 3-hexylthiophene rings (bithiophene, 2T) in an average of one of approximately five repeat units. 1H NMR spectroscopy, ultraviolet-visible spectroscopy, and gel-permeation chromatography confirm the presence and covalent incorporation of the 2T units within the conjugated backbone of the segmented polymer. The tensile modulus of the segmented polymer, 0.93 ± 0.16 GPa, is lower than that of the homopolymer, 2.17 ± 0.35 GPa. When blended with PC61BM, the segmented material produces devices with power conversion efficiencies of 2.82 ± 0.28%, which is similar to that of PDPP2FT, 2.52 ± 0.34%. These results suggest that it is possible to increase the mechanical resiliency of semiconducting polymers for solar cells without having a deleterious effect on the photovoltaic properties.
Introduction
Mechanical compliance of organic electronic devices is typically regarded as a solved—or never extant—problem, and thus the mechanical properties of modern (i.e., low-bandgap, high mobility, and high photovoltaic efficiency) conjugated polymers are generally unreported.1 Typical thicknesses of active materials (∼100 nm) and substrates (∼100 μm and recently ∼1 μm) can accommodate small bending radii without imposing significant tensile deformations to the active materials.2 Reports of ultra-flexible devices have enabled “imperceptible” electronics and skin-like devices on thin plastic foils and demonstrations of ultrathin organic solar cells with the highest power-to-mass ratio of any photovoltaic technology.3 Implementation of this technology for large-area applications and full exploitation of the benefit provided by thinness2 (including possible reductions in balance of systems costs) requires that the active materials accommodate at least modest tensile strains reversibly. Mechanical robustness is prerequisite for thinness because small environmental forces will produce large strains on ultra-thin substrates.4,5 The mechanical properties of organic semiconductors, however, exhibit a range of tensile moduli and propensity to fracture.1,6–8 Establishing not only the structural parameters that control the mechanical properties but simple methods to tune the elasticity without adversely affecting the electronic properties would be a significant benefit to the field of organic electronics.9 The establishment of such knowledge might enable truly “rubber” semiconductors, which could have a range of applications in devices for energy and biomedicine.10–12Our laboratory has studied the mechanical properties of regioregular poly(3-alkylthiophene) (P3AT) as a function of the length of the alkyl solubilizing group.1 Our observations led us to conclude that this structural element had a drastic effect on both the mechanical and photovoltaic properties.1 In particular, we concluded that the length of the side chain was inversely correlated with photovoltaic efficiency for P3AT:PC61BM, from A = hexyl to A = dodecyl, but that the length of the side chain was directly correlated with compliance.13 The tensile modulus of P3HT was nearly an order of magnitude greater (1.09 GPa) than that of P3OT (0.15 GPa), but the photovoltaic efficiency of P3HT-based devices was noted by us and others to be significantly greater than that of P3OT-based devices.13 There is a notion that electronic and mechanical properties tend to be in competition (if one places value on elasticity and ductility). Notably, Awartani et al. have shown that increasing order in the pure P3HT phases in P3HT:PC61BM blends with decreasing rate of evaporation of solvent during spin coating produces efficient—but stiff and brittle—photovoltaic active layers.6
While the regioregular P3ATs represent an important class of materials for fundamental studies of mechanical properties, it seems likely that a low-bandgap, donor–acceptor copolymer will emerge as the preferred “p-type” material,14 with a fullerene or another polymer as the “n-type” material, provided both materials can be manufactured at scale with low cost and with low environmental impact.15 To this end, a previous report measured the tensile moduli of PDPP2T-TT and PDPP2T-2T and attributed the slightly lower tensile modulus of PDPP2T-2T (0.74 GPa) to that of PDPP2T-TT (0.99 GPa) to the relative stiffness7 of the fused thienothiophene (TT) unit to that of the separated bithiophene (2T) unit (see Fig. 1 for structures).16 These values of modulus, however, are very close, and it does not seem that replacement of fused rings for isolated rings will be the most effective strategy to provide improvements in mechanical properties. Within classes of similar materials, the mechanical compliance is inversely correlated to the crystallinity.7 This effect has been noted in both P3ATs with different side chain lengths1 and P3ATs compared to highly crystalline annealed films of PBTTT.7,17 While it has previously been believed that high crystallinity was necessary for high charge transport, PDPP2T-TT exhibits balanced electron and hole mobilities for field-effect transistors that are among the highest of any material yet reported,18,19 but it is significantly less crystalline than are annealed films of PBTTT.20 Indeed, while the power conversion efficiencies (PCEs) of blends of MEH-PPV and MDMO-PPV21 with PC61BM are no longer state-of-the-art, the efficiencies are not drastically lower than that of the typical P3HT:PC61BM cell22 (∼2 times lower), even though P3HT is semicrystalline and MEH-PPV and MDMO-PPV are amorphous.23 PCDTBT is another example of a predominantly amorphous polymer24 that has achieved values of PCE in blends with PC71BM greater than typical values for P3HT:PC61BM.25,26 Thus an effective strategy to combine mechanical compliance and photovoltaic efficiency might include the use of a conjugated polymer with good transport along the molecular axis but with a disrupted ability to form large crystallites in the solid state27 which may stiffen the film.7
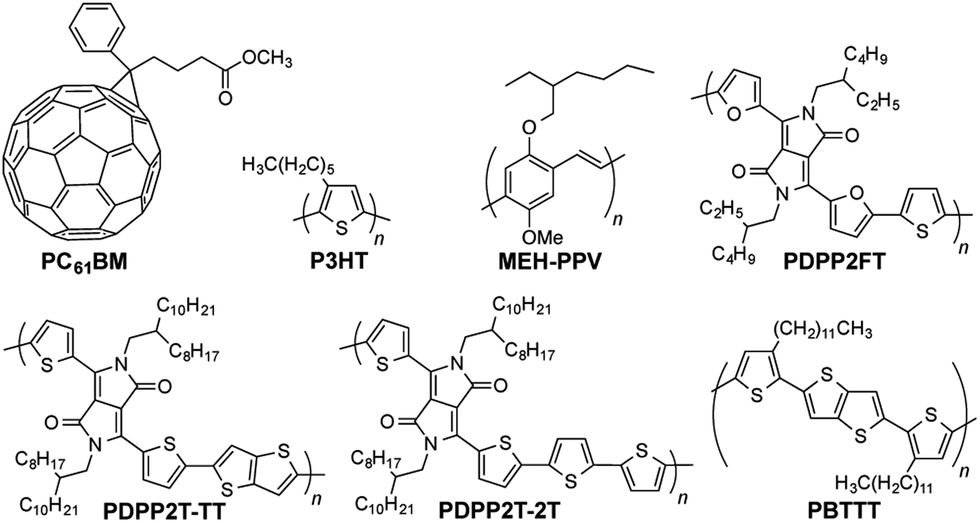 | ||
| Fig. 1 Chemical structures of materials discussed in the text. | ||
Block copolymers prepared by controlled living radical polymerization offer opportunities to combine advantageous properties of their component blocks,28 but the method is not amenable to the preparation of low-bandgap conjugated polymers. Recently, segmented, or “blocky” copolymers have been prepared by metal mediated olefin polymerization29 and also by polycondensation reactions.30 This work has demonstrated that segmented polymers can separate into domains rich in their component segments; segmentation thus provides a route to tailor the properties in a way that is analogous to block copolymerization, specifically for improved mechanical properties and processing behavior. All-conjugated block copolymers, such as analogues of regioregular polythiophenes, are generally synthesized by a chain-growth process. Alternating copolymerization, which is necessary to produce low-bandgap materials, follows step-growth kinetics and is not easily adapted to the production of block copolymers.31 Ku et al., however, recently demonstrated a hybrid strategy in which a low-bandgap copolymer was appended to a polythiophene segment bearing a reactive chain end.32 Our goal was thus to apply the strategy of segmented polymerization to a wholly low-bandgap conjugated polymer.
We focused our efforts on PDPP2FT and derivatives thereof. PDPP2FT, first reported by Woo et al., is a furan-containing donor–acceptor copolymer that is promising for photovoltaic applications.33 It is synthesized by a metal-mediated polycondensation reaction of two monomers: the DPP unit flanked by two furan rings terminated in bromides and a unit of distannylated thiophene.33 Superior solubility of polymers containing the furan moiety permits the use of ethylhexyl solubilizing groups whereas an analogous material in which the furans are substituted with thiophenes requires the much longer octyldodecyl side chains to afford useful solubility.33 Solar cells based on PDPP2FT:PC71BM blends spin-coated from chlorobenzene with a chloronapthalene additive exhibited photovoltaic efficiencies of 5.0%.33 Using PDPP2FT as a starting point, we tested a simple method for increasing the elasticity of the material through random segmentation—that is, random incorporation of an alkylated conjugated units throughout the backbone (Fig. 2). We believed this approach would have two effects: (1) disruption of the regular order in the main chain of the polymer and (2) alteration of the distribution of side chains. We predicted that both effects could lower the tensile modulus without significantly affecting the photovoltaic response of these materials in blends with fullerenes.
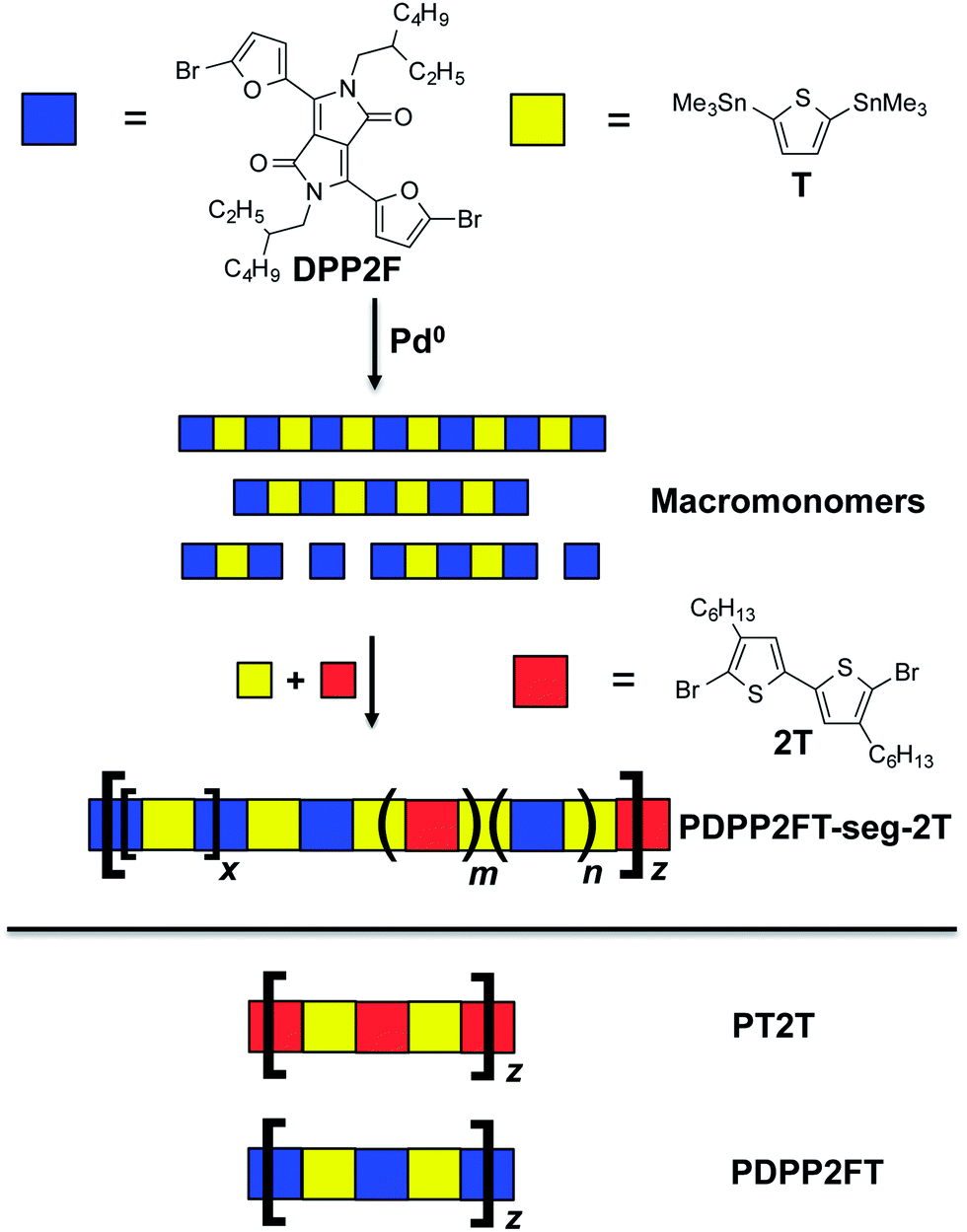 | ||
| Fig. 2 Summary of the synthetic strategy used to generate segmented copolymers. Two monomers, the dibromide (DPP2F) and the distannane (T), are reacted in the presence of Pd0. Shortly after initialization of the reaction (when “macromonomers” began to form), additional T and dibrominated bithiophene (2T) were added to the reaction mixture to form the segmented polymer, PDPP2FT-seg-2T. Separately, the homopolymers PT2T and PDPP2FT were also prepared. | ||
Experimental section
Materials
A soluble fullerene derivative, [6,6]-phenyl C61 butyric acid methyl ester (PC61BM) was obtained from Sigma-Aldrich with >99% purity. PDMS, Sylgard 184 (Dow Corning), was prepared according to the manufacturer's instructions at a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (base:crosslinker) and cured at room temperature for 36 to 48 hours before it was used for mechanical testing. (Tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane (FOTS) was obtained from Gelest. PEDOT:PSS (Clevios PH 1000) was purchased from Heraeus. DMSO was purchased from BDH with purity of 99.9% and Zonyl (FS-300) fluorosurfactant was purchased from Sigma-Aldrich.
:1 (base:crosslinker) and cured at room temperature for 36 to 48 hours before it was used for mechanical testing. (Tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane (FOTS) was obtained from Gelest. PEDOT:PSS (Clevios PH 1000) was purchased from Heraeus. DMSO was purchased from BDH with purity of 99.9% and Zonyl (FS-300) fluorosurfactant was purchased from Sigma-Aldrich.
General
All reagents were obtained from commercial suppliers and used without purification. Chloroform, ortho-dichlorobenzene (ODCB), dimethylformamide (DMF) and tetrahydrofuran (THF) were obtained from Sigma-Aldrich. All compounds were characterized by 1H NMR and 13C NMR (300 MHz, Varian) using CDCl3 as the solvent. The residual chloroform peak at 7.26 ppm was used to calibrate the chemical shifts for 1H NMR. Gel-permeation chromatography (GPC) was performed in chloroform on a Waters 2690 Separation Module equipped with a Waters 2414 Refractive Index Detector and a Waters 2996 Photodiode Array Detector. Molecular weights were calculated relative to linear PS standards. Atomic force microscope (AFM) images were obtained with a Veeco Scanning Probe Microscope in tapping mode. AFM data was analyzed with NanoScope Analysis v1.40 software (Bruker Corp.). Ultraviolet-visible (UV-vis) spectra were obtained of the polymers in chloroform and in the solid state, as-cast from 4:1 chloroform:ODCB (by volume, 5 mg mL−1) using a Perkin Elmer Lambda 1050 UV-vis-NIR spectrophotometer. We synthesized the two known polymers, PDPP2FT33 and PT2T (formerly called C6-TT),34,35 according to previously established procedures.
Synthesis of PDPP2FT-seg-2T
We synthesized this material using a method related to that of PDPP2FT, except that after allowing the DPP2F and T (Fig. 2) to react for a short time, we added brominated bithiophene monomer (2T) and additional stannylated thiophene (T), as follows. In a 12 mL reaction tube, DPP2F (234 mg, 0.360 mmol), 2,5-bis(trimethylstannyl)-thiophene (T, 147 mg, 0.360 mmol), Pd2(dba)3 (2 mol%) and P(o-tol)3 (8 mol%) were dissolved in 4 mL chlorobenzene and degassed by bubbling argon through the mixture for 20 min. In a separate identical reaction tube, 2,5-bis(trimethylstannyl)-thiophene (T, 49 mg, 0.120 mmol) and brominated bithiophene (2T, 59 mg, 0.120 mmol) were dissolved in 2 mL chlorobenzene and degassed in the same manner. The first reaction tube was heated in an oil bath to 110 °C for 15 min, and a color change was observed from red monomer to green/blue oligomeric species. The first tube was removed from the oil bath and allowed to cool, and then the contents of the second reaction tube were added by cannula. The reaction was again heated to 110 °C for 6 h and then was allowed to cool to room temperature and was diluted with chloroform to reduce viscosity, and was precipitated into cold methanol. The solid was collected on filter paper, which was loaded into a Soxhlet and extracted with methanol and hexanes before the segmented polymer was collected by extraction with chloroform. Concentration under reduced pressure yielded 235 mg of a dark solid. GPC analysis provided values of Mw = 55 kDa and PDI = 2.5. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.70–8.30 (br, 2H), 7.22–6.33 (br, 4H), 4.65–3.3 (br, 4H), 2.88–2.38 (br, 0.91H inferred, signal due to randomly incorporated 2T), 2.03–1.76 (br, 2H), 1.74–1.63 (br ovlp, 0.91H inferred), 1.60–1.06 (br, 16H), 1.04–0.70 (br ovlp, 13.36H inferred).Mechanical characterization
We measured the tensile modulus of each material using the mechanical buckling technique originally described by Stafford et al.36 This method has been used in various thin film systems including conjugated polymer films for heterojunction OPV devices.1,6,8,16 In brief, the films were spin-coated on passivated glass slides and transferred to poly(dimethylsiloxane) (PDMS) substrates bearing a small pre-strain. After transfer, the PDMS substrates were relaxed and the conjugated polymer film adopted sinusoidal buckles. The buckling wavelength, λb, is related to the thickness of the film, df, the tensile moduli of the film and the substrate, Ef and Es, and the Poisson's ratios of the two materials, νf and νs by the following equation: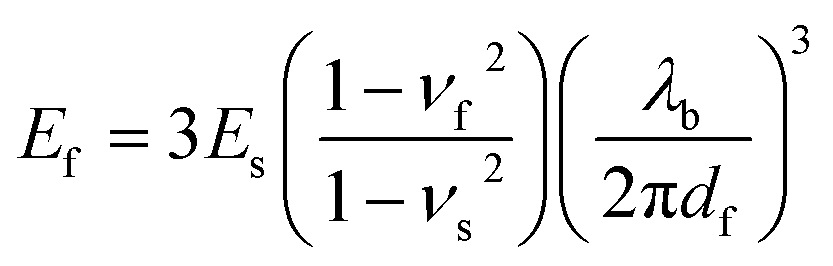 | (1) |
We measured the tensile modulus of the substrate, Es (using a commercial pull tester), the buckling wavelength, λb (by optical microscopy), and the film thickness, df (by stylus profilometry). The slope of a plot of λbvs. df for three different film thicknesses was inserted into eqn (1). The Poisson's ratios were taken as 0.5 and 0.35 for PDMS and the conjugated polymers films, which agree well with the previously reported values and our theoretical predictions.1,8
We also computed the values for the tensile moduli of the conjugated polymer using a theoretical model originally described by Seitz,37 applied to conjugated polymers by Tahk,8 and further refined by our group to account for differential glass transition temperature between various conjugated polymers.1 The model incorporated the knowledge of the chemical structure of the polymer—i.e., molecular weight, van der Waals volume, the length and the number of rotational bonds in the monomer—and the glass transition temperature (Tg).
Fabrication and testing of photovoltaic devices
The conjugated polymer:fullerene bulk heterojunction (BHJ) films were spin-coated onto glass slides pre-coated with a PEDOT:PSS films. Prior to spin-coating the PEDOT:PSS, the glass slides were cleaned with Alconox solution (2 mg mL−1), deionized water, acetone, and then isopropyl alcohol (IPA) in an ultrasonic bath for 10 min each, followed by a plasma treatment at ∼30 W for 3 min at a base pressure of 200 mTorr in ambient air. The PEDOT:PSS layer was deposited from an aqueous solution containing 93 wt% Clevios PH 1000 (∼0.9–1.2 wt% PEDOT:PSS), 6.9 wt% DMSO, and 0.1 wt% Zonyl.38 The solution was filtered with a 1 μm glass microfiber (GMF) syringe filter and then spin coated at a speed of 500 rpm (100 rpm s−1 ramp) for 60 s, followed by 2000 rpm (750 rpm s−1 ramp) for 60 s, which produced a layer 200 nm thick. The samples were subsequently dried at 150 °C for 30 min before the deposition of the polymer:fullerene BHJ films. The BHJ films were deposited from solutions of 1:2 by weight polymer and PC61BM in 4:1 chloroform:ODCB (2.5 mg mL−1), which were stirred ∼12 h and filtered with 0.20 μm poly(tetrafluoroethylene) (PTFE) syringe filters. The solutions were then spin coated onto the electrode layer at a speed of 300 rpm (100 rpm s−1 ramp) for 240 s, followed by 2000 rpm (750 rpm s−1 ramp) for 60 s. For each device, a thin strip of the PEDOT:PSS electrode was exposed by wiping away some of the polymer:PC61BM film with chloroform so that electrical contact could be made. To minimize exposure to ambient air by transferring devices into and out of an evaporator in a different building, EGaIn (extruded by hand from a syringe) was used as the top contact.39 The photovoltaic properties were measured in a nitrogen-filled glovebox using a solar simulator with a 100 mW cm−2 flux that approximated the solar spectrum under AM 1.5G conditions (ABET Technologies 11016-U up-facing unit calibrated with a reference cell with a KG5 filter). The current density versus voltage was measured for both dark and under illumination using a Keithley 2400 SourceMeter.
Results and discussion
1H NMR
Our first task was to verify the incorporation of the 2T units in the PDPP2FT-seg-2T polymer. Fig. 3 compares the 1H NMR spectra for PDPP2FT, PDPP2FT-seg-2T and PT2T; the inset highlights the signal from 3.0 to 1.5 ppm. Because the PDPP2FT and PDPP2FT-seg-2T are compositionally similar, differences in spectra were expected to be quite minor. The spectrum for PDPP2FT-seg-2T is largely similar to that of PDPP2FT, except that PDPP2FT-seg-2T exhibited a signal at 2.88–2.38 ppm and a partially overlapping signal at 1.74–1.63 ppm, which we attribute to the methylene protons located α and β to the aromatic rings of the bithiophene unit as shown in the inset of Fig. 3. From integration of the signals, we estimated that there was one 2T unit incorporated per 4.4 DPP2F units in the segmented polymer. While the 1H NMR experiments provided evidence for 2T units in our samples, further investigation was necessary to conclude that they were covalently incorporated into the main polymer chain. 13C NMR spectra of the polymer samples are shown in Fig. S1† but were inconclusive owing to a low signal to noise ratio for PDPP2FT and PDPP2FT-seg-2T, which we attribute to a low effective concentration of magnetically distinct carbon atoms even at the limit of solubility (ca. 50 mg mL−1) and with data collection times of 9 h.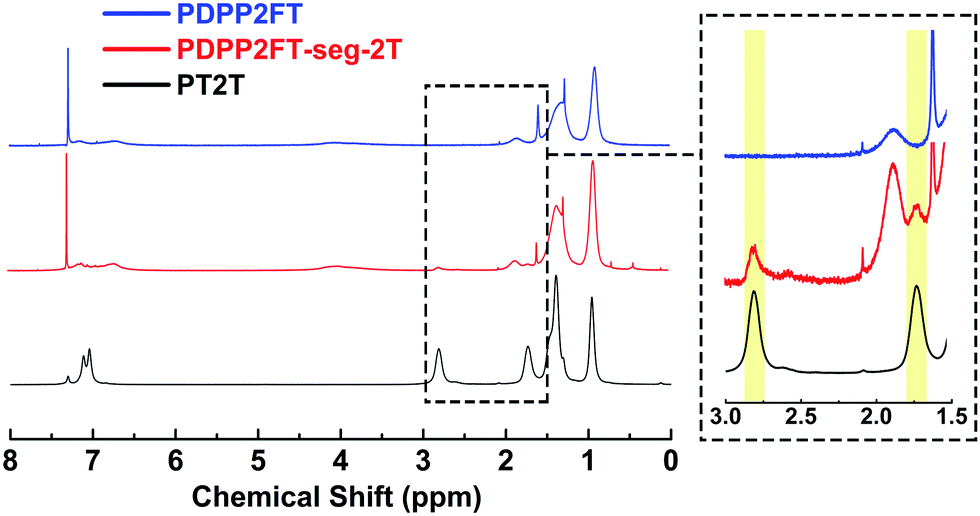 | ||
| Fig. 3 1H NMR spectra of PDPP2FT, PDPP2FT-seg-2T, and PT2T. Peaks associated with the bithiophene are highlighted in the inset. | ||
UV-visible absorption
We compared the ultraviolet-visible absorption spectra of the three materials (Fig. 4). The bandgaps were determined from the onset of absorption for thin films of the pure polymers (Fig. 4a). PT2T exhibited an onset of optical absorption at around 660 nm (bandgap = 1.88 eV), with a maximum absorption around 540 nm, while the pure PDPP2FT exhibited an onset of optical absorption at around 930 nm (bandgap = 1.33 eV), with a maximum at 800 nm. PDPP2FT-seg-2T, which contains segments of PDPP2FT interspersed by statistical incorporation of monomers (PDPP2FT-seg-2T) exhibits features similar to PDPP2FT. However, the peaks in PDPP2FT-seg-2T are broader and less defined, which could suggest decreased order from what is observed in the homopolymer, PDPP2FT. The details of the vibronic structure have been used to correlate the extent of π-stacked, ordered structures (H-aggregates) in P3HT:PC61BM blends to their tensile moduli and ductility, with samples that exhibited significant H-aggregates also exhibited increased stiffness and ductility.6 Further work would be required to correlate order as measured spectroscopically to mechanical properties for this class of low-bandgap materials.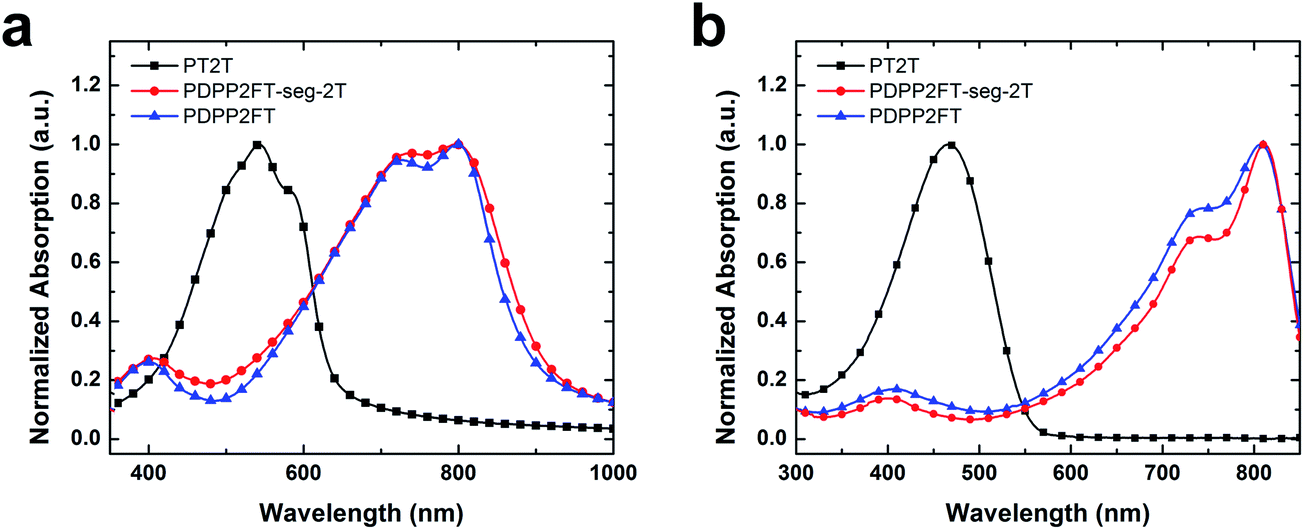 | ||
| Fig. 4 Absorption spectra of the three polymers synthesized in this work. (a) Thin films of the pure polymers spin cast from 4:1 chloroform:ODCB and (b) the pure polymers in chloroform at a concentration of 1 × 10−5 M. | ||
To determine if the bithiophene signals in the 1H NMR and the difference in absorption between the PDPP2FT and PDPP2FT-seg-2T in the UV-vis spectra were due to contamination of the PT2T homopolymer in the PDPP2FT-seg-2T sample (as opposed to covalently incorporated bithiophene units), we performed two additional UV-vis experiments. We first measured the extinction coefficients of the pure polymers from their absorption in chloroform (1 × 10−5 M) and used these values to calculate the absorption spectra of physical blends of PDPP2FT:PT2T (Fig. 4b). Because the samples were dilute, we assumed no aggregation and no interaction between unlike polymers. We thus assumed that we could treat spectra of samples containing more than one polymer as superpositions of the individual components. We then corrected for the unequal extinction coefficients of PT2T and PDPP2FT (PDPP2FT is 45% more absorbing than PT2T) and calculated the maximum ratio of PDPP2FT to PT2T that would still produce a mathematically undetectable increase in absorption in the region where PT2T absorbs most intensely, around 475 nm. We calculated this maximum ratio of PDPP2FT to PT2T to be 100:1. The normalized absorption spectra of the pure polymers and the calculated spectrum of the 100:1 physical blend, as well as the ratio of DPP2F:2T in PDPP2FT-seg-2T (4.4:1) are plotted in Fig. S2.† We then determined the absorption of a thin film of a 100:1 physical blend of PDPP2FT:PT2T (Fig. S3†). The absorption spectrum of the physical blend essentially overlapped with that of the pure PDPP2FT, except that the blend had better defined peaks than did PDPP2FT-seg-2T. These data, combined with the 1H NMR spectra, are consistent with our interpretation that the bithiophene units are covalently incorporated into the PDPP2FT-seg-2T polymer, and that the UV-vis spectra of this material does not simply represent a superposition of PDPP2FT and PT2T at either the smallest detectable amount of PT2T in PDPP2FT (100:1) or the largest possible amount based on the NMR experiments (4.4:1).
Gel-permeation chromatography
From the 1H NMR spectra, we demonstrated that both PDPP2FT and 2T units are present in the product. The next essential step was to confirm the purity—i.e., the absence of homopolymers—within the segmented product. Fig. 5 shows the GPC traces (intensity vs. retention time) and contour plots (wavelength vs. retention time) of all three conjugated polymer samples. For PDPP2FT (Fig. 5a), the main absorbance peak occurred at 550–800 nm from 10 to 14 min with a much smaller peak at 350–450 nm. Minor tailing was observed in the GPC traces; these tails probably correspond to lower molecular weight polymers. A relatively polydisperse sample was expected from a step-growth mechanism. The GPC trace for the segmented polymer (PDPP2FT-seg-2T) also showed similar tailing and a broad shoulder. This shoulder may have originated from either lower molecular weight segmented polymers or from residual homopolymers. We addressed this concern using the contour plot based on a photodiode UV detector as described by Hawker and coworkers for an all-conjugated block copolymer.32 The contour plot for the segmented polymer (PDPP2FT-seg-2T, Fig. 5b) exhibited two absorbance peaks at 350–550 and 550–800 nm centered on a retention time of 11–14 min. The plot for PT2T (Fig. 5c) also suggests the absence of major impurities; it exhibited a dominant absorbance peak from 350–500 nm at 13 min. The mechanism of polymerization suggested in Fig. 2—through chain-terminating side reactions or the formation of small concentrations of homopolymers—is consistent with some contamination by pure PT2T or PDPP2FT that were not washed out by Soxhlet extraction. The analysis by GPC, however, strongly suggests that the PDPP2FT-seg-2T sample was not contaminated by significant amounts of the PT2T homopolymer, and the statistics of polymerization suggest that PDPP2FT could only be present in very small quantities. Contamination due to homopolymers would produce two distinct absorption regions with different retention times. The closeness in retention time of PDPP2FT and PDPP2FT-seg-2T suggested a minimal difference in molecular weight (if we assume that the two structurally similar materials adopt similar conformations in solution). We thus neglected the effects of molecular weight on the mechanical properties and photovoltaic properties of the two materials (Table 1).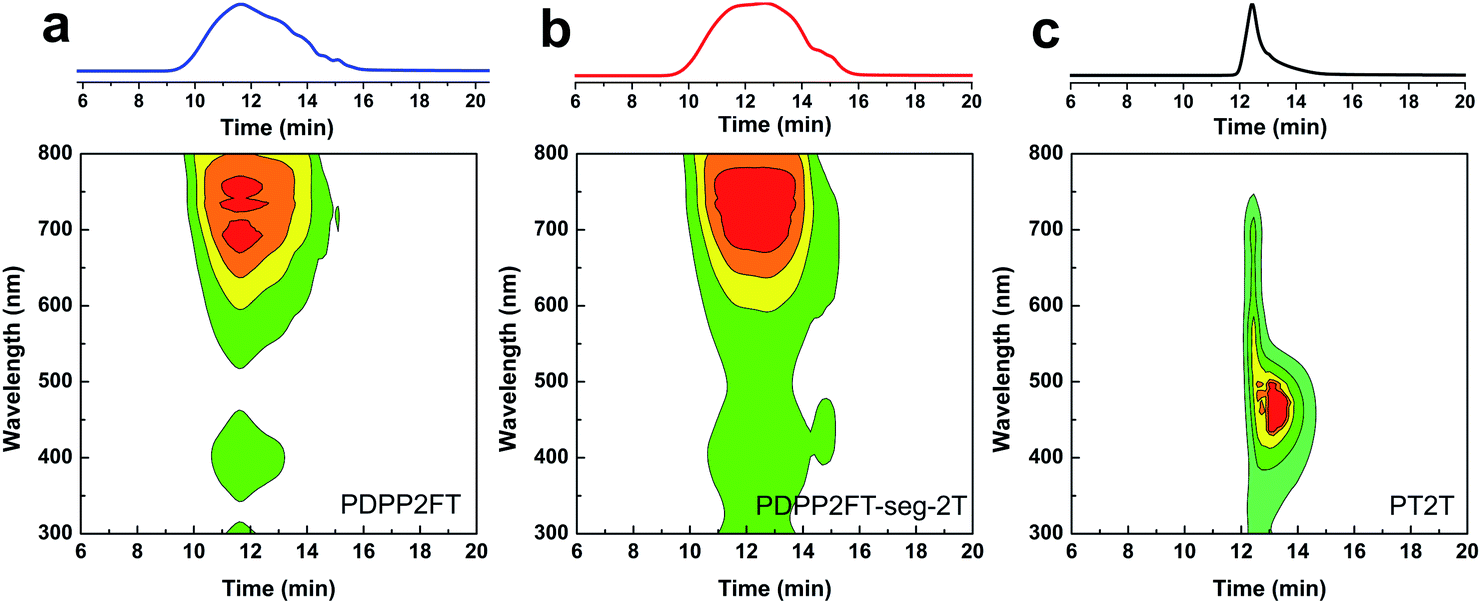 | ||
| Fig. 5 GPC traces and contour plots for (a) PDPP2FT, (b) PDPP2FT-seg-2T, and (c) PT2T based on a UV detector. | ||
| Polymer | M n (g mol−1) | M w (g mol−1) | PDI |
|---|---|---|---|
| PDPP2FT | 26400 |
69600 |
2.64 |
| PDPP2FT-seg-2T | 22300 |
55300 |
2.47 |
| PT2T | 14800 |
19400 |
1.31 |
Tensile moduli of conjugated polymer thin films
We determined the tensile moduli of the pure polymer thin films spin-coated from chloroform. For each film, the buckling wavelengths were plotted as a function of the film thickness. The slopes of the linear fits were then substituted into eqn (1) to obtain the tensile moduli of the thin films. The tensile modulus of PT2T, whose structure is closely related to P3HT,35 was determined to be 1.11 ± 0.19 GPa. This value agrees well with the values of P3HT reported previously by our group1 and literature values8,16 obtained using the same method. The obtained value for PDPP2FT, 2.17 ± 0.35 GPa, was twice that of PT2T. This value was greater than that previously reported for PDPP2T-TT (0.99 GPa),16 though we note that PDPP2T-TT contains octyldodecyl side chains and PDPP2FT contains ethylhexyl side chains. Long alkyl side chains tend to reduce the tensile modulus and increase the ductility of a conjugated polymer significantly.1We then measured the tensile modulus of the segmented polymer, PDPP2FT-seg-2T. The incorporation of the 2T units, as determined from 1H NMR spectra, produced a significantly reduced stiffness (modulus = 0.93 ± 0.16 GPa) compared to PDPP2FT. The reduction in modulus by segmentation is possibly attributable to three effects. The first possible effect is that structural randomness is correlated with decreased crystallinity, which is correlated with increased mechanical compliance in polymeric films with similar chemical structures.7 The second possible effect is that in the segmented sample, approximately one of every five DPP2F units is substituted for a 2T unit. Substitution of fused rings for isolated rings have been correlated to decreased stiffness of the film in both polythiophene7 and DPP-based systems.16 The third effect is that statistical incorporation of alkylated bithiophene units significantly altered the distribution of side chains compared to that of the homopolymer. While predicting the effect of this change in the distribution of side chains on the mechanical properties would be difficult to accomplish, small changes in the lengths of the side-chains have significant effects on the thermal, electrical, and mechanical properties of P3ATs.1,40 As a control experiment, we also measured the tensile modulus of the 100:1 physical blend between PDPP2FT and PT2T, which represents the maximum amount of contamination by PT2T homopolymer that could be undetected by UV-vis. We found that, within experimental error, the physical blend had a comparable tensile modulus to PDPP2FT (Fig. S4 and Table S1†).
Our theoretical calculations of the tensile moduli from the molecular structure of the monomer as well as the Tg of the polymer1,8,37 agreed extremely well with experimental values for the homopolymers, PT2T and PDPP2FT. The calculated values were 1.13 ± 0.14 GPa (PT2T) and 2.47 ± 0.30 GPa (PDPP2FT), using the Tg values of 14 °C and 50 °C. This simple theoretical model, however, failed to predict the reduction in modulus of PDPP2FT-seg-2T relative to that of the homopolymer, PDPP2FT. We attribute its failure primarily to its inability to incorporate the effects of randomness in the polymer chain.
Photovoltaic characteristics
To determine the applicability of these materials in organic solar cells, we fabricated devices by mixing the polymers in a 1:2 ratio with PC61BM. We used PEDOT:PSS as the transparent anode and eutectic gallium–indium (EGaIn) as the cathode.39Fig. 6 shows the current density vs. voltage (J–V) plots for representative devices (Fig. S5 and Table S2† include devices fabricated with a 100:1 PDPP2FT:PT2T physical blend, which performed slightly poorer than the PDPP2FT devices, but within error). The poor behavior we observed for the PT2T sample is consistent with similarly poor performance reported by Koppe et al.,35 who attributed the inefficiency of PT2T:PC61BM compared to P3HT:PC61BM (despite favorable offsets of the frontier molecular orbitals) to intercalation of PC61BM within the large gap between side chains in PT2T and suppression of the ability of the polymer to crystallize.35 The power conversion efficiency (PCE) of PDPP2FT:PC61BM (PCE = 2.52 ± 0.34%, N = 7) and PDPP2FT-seg-2T:PC61BM (PCE = 2.82 ± 0.28%, N = 6), however, were similar. The data for all devices tested are summarized in Table 2. The short circuit current (Jsc), open circuit voltage (Voc), fill factor (FF), and PCE are all very similar for PDPP2FT and PDPP2FT-seg-2T. The similarity in figures of merit suggests that the charge-transport properties are preserved despite the incorporation of 2T units. Interestingly, even though the incorporation of the 2T units increased the mechanical compliance of PDPP2FT-seg-2T, it did not appear to have a deleterious effect on the photovoltaic properties.
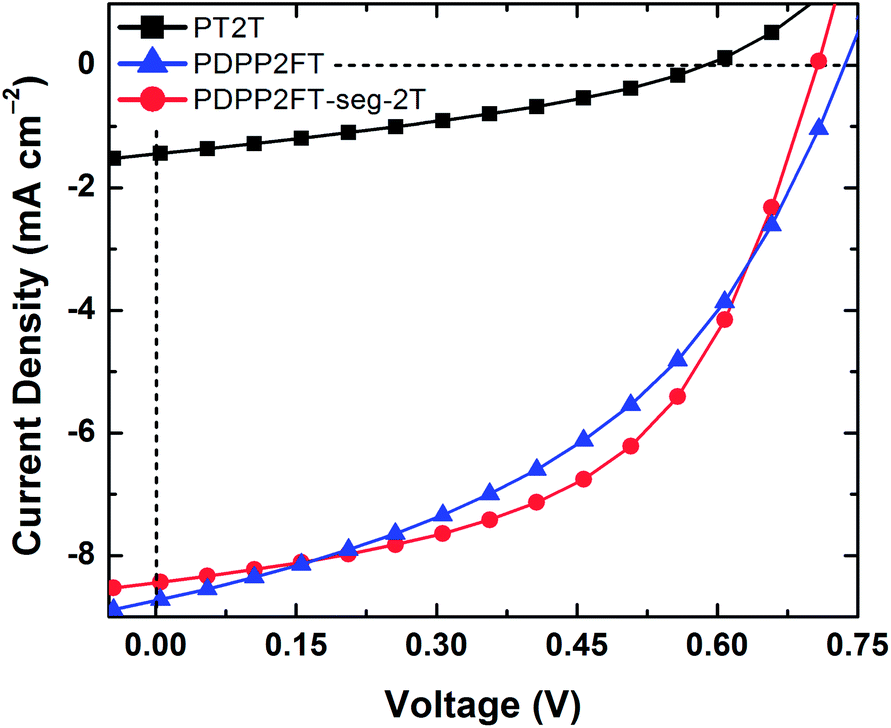 | ||
| Fig. 6 Photovoltaic characteristics of representative devices with PEDOT:PSS/polymer:PC61BM/EGaIn architecture. All active layers comprised 1:2 polymer:PC61BM spin-coated from 4:1 chloroform:ODCB. | ||
| n | J sc [mA cm−2] | V oc [mV] | FF [%] | η e [%] | |
|---|---|---|---|---|---|
| PT2T | 3 | 1.5 ± 0.1 | 579 ± 21 | 32.9 ± 1.1 | 0.28 ± 0.01 |
| PDPP2FT-seg-2T | 6 | 8.4 ± 0.5 | 699 ± 23 | 48.2 ± 3.3 | 2.82 ± 0.28 |
| PDPP2FT | 7 | 8.3 ± 0.5 | 715 ± 25 | 42.5 ± 3.6 | 2.52 ± 0.34 |
Atomic force microscopy
To determine if the differences in tensile moduli of PDPP2FT and PDPP2FT-seg-2T could be attributed to a significant change in the morphology of the films, we examined spin-coated films by AFM. Previous studies have suggested that roughness observable by AFM correlates with crystalline order, as determined by grazing-incidence X-ray diffraction, in conjugated polymer films annealed below Tm.41 A similar effect was noted in a series of P3ATs from A = butyl to A = dodecyl, where the shortest alkyl chains had the greatest roughness (presumably due to greater crystallinity) and stiffness.1Fig. 7 shows AFM micrographs of the heights of the PDPP2FT and the PDPP2FT-seg-2T films. We observed that PDPP2FT had a root mean square (rms) roughness of 1.13 ± 0.09 nm and PDPP2FT-seg-2T had an rms roughness of 1.25 ± 0.08 nm. The similarity of these values suggests that the correlation between roughness (as a manifestation of crystallinity) and tensile modulus—as observed in other systems—is not general. | ||
| Fig. 7 Height images from atomic force micrographs of unannealed polymer thin films. (a) PDPP2FT and (b) PDPP2FT-seg-2T. | ||
Competition between photovoltaic performance and stiffness
Within groups of structurally related conjugated polymers, charge transport and photovoltaic efficiency are regarded as antithetical to mechanical compliance.1,6,7,13 Along with the tensile moduli of the pure polymers, we measured the moduli of the 1:2 polymer:PC61BM blends spin-coated from 4:1 chloroform:ODCB. Fig. 8a compares the tensile moduli of the pure polymer films and the blended films. For all three polymers, we observed an increase in the tensile moduli with the addition of PC61BM. Various studies have reported the same trend in systems comprising conjugated polymer and fullerene composites.1,7,8,13,16 In Fig. 8b, we plotted the power conversion efficiencies (PCE) of the polymer:PC61BM BHJ films as a function of tensile modulus. For materials in which these figures of merit are strongly correlated, such as in P3HT:PC61BM exhibiting increasing order, the data points would sit (very roughly) on a diagonal extending from low tensile modulus and low PCE to high values of both parameters. Interestingly, the sample PDPP2FT-seg-2T:PC61BM shows a similar PCE to that of PDPP2FT:PC61BM, but the PDPP2FT-seg-2T is a factor of two more elastic than PDPP2FT. While polymer:PC61BM blends are always measured to be stiffer than the pure polymers, the factor by which the tensile modulus of the blend is greater than that of the pure polymer tends to be similar within similar classes of materials.1,16 The segmented copolymer, PDPP2FT-seg-2T appears to exhibit photovoltaic properties resembling PDPP2FT, but mechanical properties resembling those of PT2T. It is possible that the “random” segments interspersed between PDPP2FT segments have a softening effect on the material. The all-conjugated nature of the “random” segments may provide advantages over block copolymers in which the plasticizing block is insulating.42
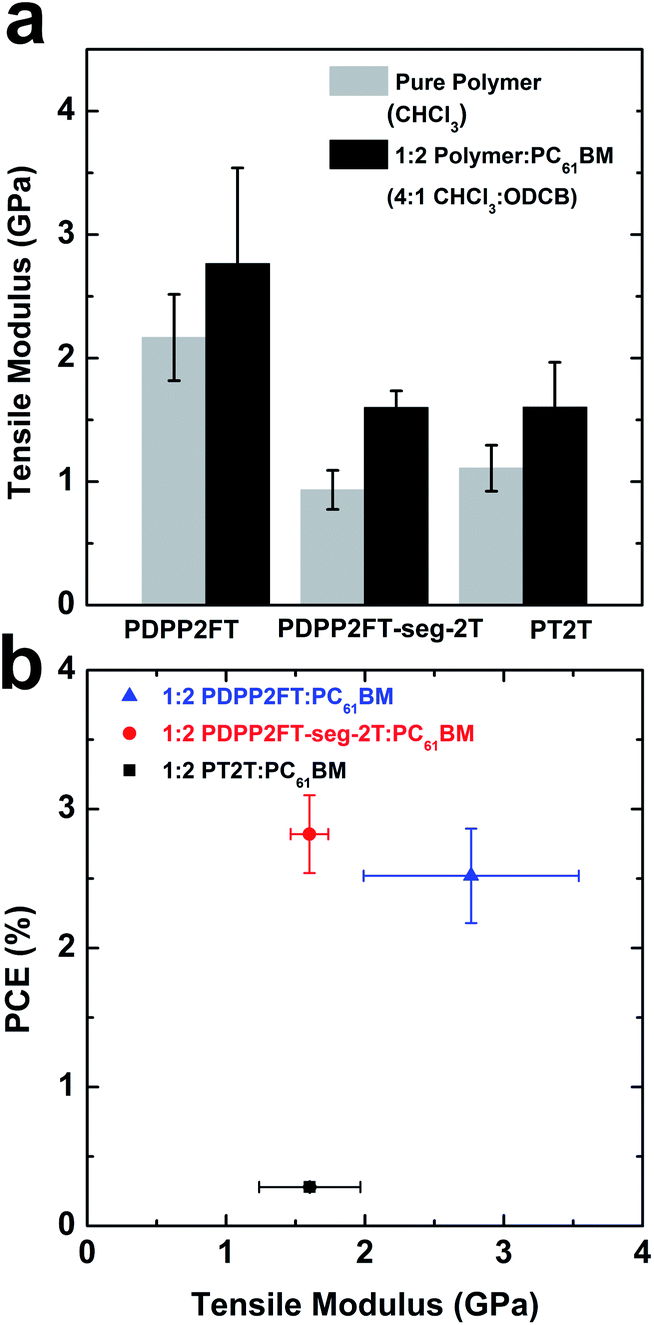 | ||
| Fig. 8 Mechanical properties of pure polymers and polymer:fullerene composites. (a) Comparison between the tensile moduli of pure polymer films spin-coated from chloroform and the films comprising 1:2 polymer:PC61BM blends spin-coated from 4:1 chloroform:ODCB. (b) Plot of power conversion efficiency of the polymers in a 1:2 blend with PC61BM spin-coated from 4:1 chloroform:ODCB. The architecture of the devices was PEDOT:PSS/polymer:PC61BM/EGaIn. The vertical error bars for 1:2 PT2T:PC61BM sample overlap with the marker. | ||
Conclusion
We have demonstrated that segmentation could be an effective strategy to increase the mechanical compliance of low-bandgap conjugated polymers without deleteriously affecting their optoelectronic properties. The method does not add significant complexity to the synthetic protocol—a third monomer is simply added to the reaction mixture after a predetermined length of time. The polymerization strategy described here suggests the possibility of fully segmented polymers comprising “macromonomers” of polymers with different band structures. Such materials could be analogous to block copolymers except that block copolymers are synthesized by living, chain-growth processes. In contrast, polymers in which both components are synthesized by step-growth processes (i.e., the Stille polymerization) are not amenable to the synthesis of block copolymers. Segmented polymerization may therefore be a route to synthesizing organic semiconductors with tailored thermal and mechanical properties (i.e., semiconducting thermoplastic elastomers). Our analysis also exposed deficiencies in the ways in which standard semi-empirical theories predict mechanical properties in semicrystalline polymers. Future work will attempt to incorporate the effects of randomness in the polymer backbones as well as address the behavior of these softened polymers in real-world conditions.Acknowledgements
This work was supported by the Air Force Office of Scientific Research (AFOSR) Young Investigator Program, grant number FA9550-13-1-0156. Additional support was provided by the NSF Graduate Research Fellowship under grant number DGE-1144086, awarded to S. S., and by laboratory startup funds from the University of California, San Diego.References
- S. Savagatrup, A. S. Makaram, D. J. Burke and D. J. Lipomi, Adv. Funct. Mater., 2014, 24, 1169–1181 CrossRef CAS.
- M. Kaltenbrunner, M. S. White, E. D. Glowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 770 CrossRef PubMed.
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwodiauer, I. Graz, S. Bauer-Gogonea, S. Bauer and T. Someya, Nature, 2013, 499, 458–463 CrossRef CAS PubMed.
- S. R. Dupont, M. Oliver, F. C. Krebs and R. H. Dauskardt, Sol. Energy Mater. Sol. Cells, 2012, 97, 171–175 CrossRef CAS PubMed.
- V. Brand, C. Bruner and R. H. Dauskardt, Sol. Energy Mater. Sol. Cells, 2012, 99, 182–189 CrossRef CAS PubMed.
- O. Awartani, B. Lemanski, H. W. Ro, L. J. Richter, D. M. DeLongchamp and B. T. O'Connor, Adv. Energy Mater., 2013, 3, 399–406 CrossRef CAS.
- B. O'Connor, E. P. Chan, C. Chan, B. R. Conrad, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, C. L. Soles and D. M. DeLongchamp, ACS Nano, 2010, 4, 7538–7544 CrossRef PubMed.
- D. Tahk, H. H. Lee and D. Y. Khang, Macromolecules, 2009, 42, 7079–7083 CrossRef CAS.
- D. J. Lipomi and Z. N. Bao, Energy Environ. Sci., 2011, 4, 3314–3328 CAS.
- T. Sekitani and T. Someya, MRS Bull., 2012, 37, 236–245 CrossRef.
- J. A. Rogers, T. Someya and Y. G. Huang, Science, 2010, 327, 1603–1607 CrossRef CAS PubMed.
- D. H. Kim, N. S. Lu, R. Ma, Y. S. Kim, R. H. Kim, S. D. Wang, J. Wu, S. M. Won, H. Tao, A. Islam, K. J. Yu, T. I. Kim, R. Chowdhury, M. Ying, L. H. Xu, M. Li, H. J. Chung, H. Keum, M. McCormick, P. Liu, Y. W. Zhang, F. G. Omenetto, Y. Huang, T. Coleman and J. A. Rogers, Science, 2011, 333, 838–843 CrossRef CAS PubMed.
- T. F. O'Connor, A. V. Zaretski, B. A. Shiravi, S. Savagatrup, A. D. Printz, M. I. Diaz and D. J. Lipomi, Energy Environ. Sci., 2014, 7, 370–378 Search PubMed.
- L. T. Dou, J. B. You, Z. R. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed.
- D. J. Burke and D. J. Lipomi, Energy Environ. Sci., 2013, 6, 2053–2066 CAS.
- D. J. Lipomi, H. Chong, M. Vosgueritchian, J. G. Mei and Z. N. Bao, Sol. Energy Mater. Sol. Cells, 2012, 107, 355–365 CrossRef CAS PubMed.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. M. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed.
- Z. Y. Chen, M. J. Lee, R. S. Ashraf, Y. Gu, S. Albert-Seifried, M. M. Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Mater., 2012, 24, 647–652 CrossRef CAS PubMed.
- H. Bronstein, Z. Y. Chen, R. S. Ashraf, W. M. Zhang, J. P. Du, J. R. Durrant, P. S. Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed.
- X. R. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073–15084 CrossRef CAS PubMed.
- S. E. Shaheen, C. J. Brabec, N. S. Sariciftci, F. Padinger, T. Fromherz and J. C. Hummelen, Appl. Phys. Lett., 2001, 78, 841–843 CrossRef CAS PubMed.
- M. Jorgensen, J. E. Carle, R. R. Sondergaard, M. Lauritzen, N. A. Dagnaes-Hansen, S. L. Byskov, T. R. Andersen, T. T. Larsen-Olsen, A. P. L. Bottiger, B. Andreasen, L. Fu, L. J. Zuo, Y. Liu, E. Bundgaard, X. W. Zhan, H. Z. Chen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2013, 119, 84–93 CrossRef PubMed.
- T. Qin and A. Troisi, J. Am. Chem. Soc., 2013, 135, 11247–11256 CrossRef CAS PubMed.
- A. J. Pearson, T. Wang, A. D. F. Dunbar, H. N. Yi, D. C. Watters, D. M. Coles, P. A. Staniec, A. Iraqi, R. A. L. Jones and D. G. Lidzey, Adv. Funct. Mater., 2013, 24, 659 CrossRef.
- J. H. Seo, A. Gutacker, Y. M. Sun, H. B. Wu, F. Huang, Y. Cao, U. Scherf, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 8416–8419 CrossRef CAS PubMed.
- T. P. Osedach, T. L. Andrew and V. Bulovic, Energy Environ. Sci., 2013, 6, 711–718 CAS.
- J. Rivnay, M. F. Toney, Y. Zheng, I. V. Kauver, Z. H. Chen, V. Wagner, A. Facchetti and A. Salleo, Adv. Mater., 2010, 22, 4359–4363 CrossRef CAS PubMed.
- C. J. Hawker and T. P. Russell, MRS Bull., 2005, 30, 952–966 CrossRef CAS.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 213, 714–719 CrossRef PubMed.
- M. Zhang, R. B. Moore and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3710–3718 CrossRef CAS.
- U. Scherf, A. Gutacker and N. Koenen, Acc. Chem. Res., 2009, 41, 1086–1097 CrossRef PubMed.
- S. Y. Ku, M. A. Brady, N. D. Treat, J. E. Cochran, M. J. Robb, E. Kramer, M. L. Chabinyc and C. J. Hawker, J. Am. Chem. Soc., 2012, 134, 16040–16046 CrossRef CAS PubMed.
- C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 15547–15549 CrossRef CAS PubMed.
- S. Tierney, M. Heeney and I. McCulloch, Synth. Met., 2005, 148, 195–198 CrossRef CAS PubMed.
- M. Koppe, M. Scharber, C. J. Brabec, W. Duffy, M. Heeney and I. McCulloch, Adv. Funct. Mater., 2007, 17, 1371–1376 CrossRef CAS.
- C. M. Stafford, C. Harrison, K. L. Beers, A. Karim, E. J. Amis, M. R. Vanlandingham, H. C. Kim, W. Volksen, R. D. Miller and E. E. Simonyi, Nat. Mater., 2004, 3, 545–550 CrossRef CAS PubMed.
- J. T. Seitz, J. Appl. Polym. Sci., 1993, 49, 1331–1351 CrossRef CAS.
- M. Vosgueritchian, D. J. Lipomi and Z. N. Bao, Adv. Funct. Mater., 2012, 22, 421–428 CrossRef CAS.
- A. Du Pasquier, S. Miller and M. Chhowalla, Sol. Energy Mater. Sol. Cells, 2006, 90, 1828–1839 CrossRef CAS PubMed.
- A. Babel and S. A. Jenekhe, Synth. Met., 2005, 148, 169–173 CrossRef CAS PubMed.
- E. Verploegen, R. Mondal, C. J. Bettinger, S. Sok, M. F. Toney and Z. Bao, Adv. Funct. Mater., 2010, 20, 3519–3529 CrossRef CAS.
- C. Muller, S. Goffri, D. W. Breiby, J. W. Andreasen, H. D. Chanzy, R. A. J. Janssen, M. M. Nielsen, C. P. Radano, H. Sirringhaus, P. Smith and N. Stingelin-Stutzmann, Adv. Funct. Mater., 2007, 17, 2674–2679 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00029c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2014 |