
Self-assembled ultra-small zinc stannate nanocrystals with mesoscopic voids via a salicylate templating pathway and their photocatalytic properties
Abstract
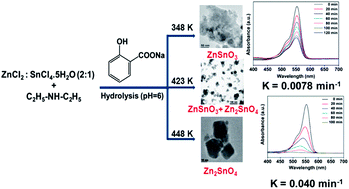
Self-assembled, tiny zinc stannate nanocrystals with controlled crystalline phases of ZnSnO3 (MZS-1) and Zn2SnO4 (MZS-2) together with mesoscopic void spaces have been synthesized via a salicylate templating method with particle dimensions of ca. 1–2 nm and 17–20 nm for MZS-1 and 2, respectively. These nanomaterials have been thoroughly characterized by powder XRD, HR TEM, N2-sorption, TG-DTA and UV-visible spectroscopic tools. Self-aggregation of the tiny nanoparticles could be responsible for these interparticle mesopores in these zinc stannate nanocrystals. The Zn2SnO4 nanomaterial showed higher photocatalytic activity over that of ZnSnO3 in the photodegradation of Rhodamine B under the irradiation of UV/visible light.
 Please wait while we load your content...
Please wait while we load your content...

