
Facile synthesis of well-dispersed Pd–graphene nanohybrids and their catalytic properties in 4-nitrophenol reduction†
Abstract
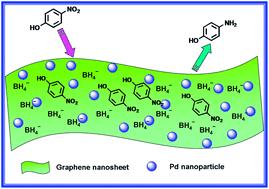
In this work, we described a simple and effective synthesis method for preparing well-dispersed Pd–graphene nanohybrids through simultaneous chemical reduction of functionalized graphene oxide and Pd2+ ions. The structure and physicochemical properties of the resulting nanohybrids were characterized in detail by Fourier transformation infrared spectroscopy, Raman, X-ray photoelectron spectroscopy, high-resolution transmission electron microscopy and scanning electron microscopy. The obtained results indicated that Pd nanoparticles distributed uniformly on the surface of the functionalized graphene when the loading amount of Pd is less than 1.0 wt%. Moreover, the so-formed Pd-FG hybrid material could be well dispersed in water, forming a homogeneous dispersion. This nanohybrid, combining the unique catalytic properties of Pd with the excellent adsorption and electron transfer ability of graphene, exhibited enhanced catalytic activity toward the reduction of 4-nitrophenol by NaBH4 even under lower Pd loading.
 Please wait while we load your content...
Please wait while we load your content...

