 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zeolites and related sorbents with narrow pores for CO2 separation from flue gas
Ocean Cheung
and
Niklas Hedin
*
Department of Materials and Environmental Chemistry, Berzelii Center EXSELENT on Porous Materials, Arrhenius Laboratory, Stockholm University, S-106 91, Stockholm, Sweden. E-mail: niklas.hedin@mmk.su.se
First published on 7th March 2014
Abstract
Adsorbents with small pores are especially relevant for capturing carbon dioxide at large emission sources. Such sorbents could be used potentially to reduce the energy demands for separating carbon dioxide from flue gas as compared with today's technologies. Here, we review the literature for crystalline, inorganic, and potentially inexpensive adsorbents. A number of different adsorbents with narrow pore openings are compared.
 Ocean Cheung | Ocean Cheung, graduated (MChem) from the University of Warwick, UK, in 2009. He started his PhD at the Department of Materials and Environmental Chemistry at Stockholm University, Sweden, in 2009. He has been working on zeolites and zeolite-like adsorbents for CO2 separation. |
 Niklas Hedin | Niklas Hedin, PhD from the Royal Institute of Technology, Stockholm, Sweden. Post doc with Bradley Chmelka in UC California at Santa Barbara, and with Sebastian Reyes at ExxonMobil in Annadale, US. Now Associate Professor in Materials Chemistry and Director for the Berzelii Center EXSELENT, Department of Materials and Environmental Chemistry at Stockholm University, Sweden. |
General introduction
Adsorption-driven capture of CO2 from flue gas or natural gas is currently investigated as a potential replacement for absorption processes.1 For carbon capture and storage (CCS), adsorption-driven capture could ideally reduce the cost for capture of CO2.2 The high cost for the capture step of CCS is one of the reasons why it has not been implemented yet. Even though, this review focuses on capture of CO2 from N2-rich mixtures, several of the sorbents are relevant for natural gas and biogas upgrading as well.Adsorbents introduction
Several sorbents classes have been investigated as CO2 sorbents and include large pore zeolites, metal organic frameworks, amine modified silica materials. Recent reviews of these sorbents include Choi et al.,1 Li et al.3 and Moliner et al.4 In particular, inorganic sorbents with narrow pore openings have advantages when it comes to selectivity for CO2, uptake of CO2, stability, and potential cost. Crystalline porous sorbents of the zeolitic kind with narrow pore windows are defined specifically as such compounds with a primary pore window opening encircled by 8 oxygen atoms. Such zeolite-type materials are classified as 8-ring zeolites. The narrow pore windows are of interest because their overall pore dimensions falls close to the effective kinetic diameters of CO2 and N2. It is important to note that the effective kinetic diameter of CO2 is smaller than that of N2 within porous solids, in contrast to the diameters in gaseous state. Typical values of the effective kinetic diameters within zeolites are 0.33 nm for CO2 and 0.36 nm for N2.5 Effective kinetic diameters here refer to the minimum diameters of CO2 and N2 in a porous solid, these quantities will be referred to throughout this review. Notably, these gases have larger molecular diameters in gas phase. In gas phase, CO2 (0.51 nm) has a larger diameter than N2 (0.43 nm).6The CO2-over-N2 selectivity of a sorbent can have thermodynamic, kinetic and possibly molecular sieving contributions. Thermodynamic contributions towards CO2 selectivity are related to the significantly lower temperature of condensation (or boiling) for N2 (77 K) as compared with the solidification (or sublimation) temperature of CO2 (194 K). Furthermore, CO2 also has a higher quadrupole moment (−13.7 × 10−24 cm2) than N2 (−4.9 × 10−26 cm2). Hence, CO2 interacts more significantly with the electrical field gradients of the sorbents (such as zeolites) than N2. It is also important to note that neither CO2 nor N2 have dipole moments. The lack of dipole moments means that the interaction between CO2 or N2 and the framework's electrical field is not related to permanent dipole moments, but rather to the polarizability of CO2 and N2. The kinetic contribution towards selectivity is related to a reduced N2 diffusivity. N2 diffusivity can become very low when the size of the pore window aperture approaches the effective kinetic diameter of N2. For such cases, N2 will be effectively eliminated from sorption when the uptake rate is distinctly slower than the characteristic time of the adsorption process. CO2 on the other hand due to the smaller kinetic diameter, will sense less restriction on its diffusion throughout the pores of sorbents with narrow pore windows. Under such circumstances kinetics and possibly molecular sieving would contribute to an enhanced CO2-over-N2 selectivity. The CO2-over-N2 selectivity of different sorbents can be compared by calculating the separation factor (s). This factor (s) is defined as:
| s = (q1/q2)/(p1/p2) |
Flue gas from a coal burning power plant typically contains up to 15 vol% of CO2 (the rest being mainly N2).7 In this review we consider a hypothetical flue gas stream which has a pressure feed of 100 kPa and contains 15 vol% CO2 (p1 = 15 kPa), 85 vol% N2 (p2 = 85 kPa). The CO2 uptake of the different sorbents (273 K unless otherwise stated) at 15 kPa and N2 uptake at 85 kPa are listed in Table 1.
| Adsorbent | CO2 uptake at 0.15 bar (mmol g−1) | N2 uptake at 0.85 bar (mmol g−1) | Selectivity (s) | Ref. |
|---|---|---|---|---|
| K-CHA | 4.0 | 0.85 | 27 | 27 |
| Na-CHA | 4.2 | 1.3 | 18 | 27 |
| Li-CHA | 4.4 | 0.53 | 47 | 27 |
| Ba-CHA | 3.0 | 1.1 | 15 | 27 |
| Mg-CHA | 3.4 | 0.65 | 30 | 27 |
| NaA | 3.2 | 0.30 | 60 | 28 |
| NaKA (17% K+) | 2.3 | 0.02 | 660 | 28 |
| MgA | 2.4 (298 K) | 0.25 (298 K) | 54 | 29 |
| CaA | 4.0 (298 K) | 0.50 (298 K) | 45 | 29 |
| CaA | 2.6 (303 K) | 0.20 (303 K) | 74 | 30 |
| H-RHO | 1.6 (0.1 bar, 298 K) | — | — | 31 |
| Li-RHO | 3.3 (0.1 bar, 298 K) | — | — | 31 |
| Na-RHO | 3.1 (0.1 bar, 298 K) | — | — | 31 |
| K-RHO | 1.5 (0.1 bar, 298 K) | — | — | 31 |
| Cs-RHO | 0.07 (0.1 bar, 298 K) | — | — | 31 |
| NaCs-RHO | 2.6 (283 K) | — | — | 32 |
| Zeolite T | 2.6 (298 K) | 0.40 (298 K) | 37 | 33 |
| Zeolite T | 1.8 (298 K) | 0.17 (298 K) | 60 | 34 |
| Zeolite T | 2.7 (288 K) | 0.40 (288 K) | 38 | 34 |
| H-ZK-5 | 1.1 | 0.10 | 62 | 35 |
| Li-ZK-5 | 3.9 | 0.23 | 96 | 35 |
| Na-ZK-5 | 3.4 | 0.27 | 71 | 35 |
| K-ZK-5 | 3.0 | 0.23 | 74 | 35 |
| Mg-ZK-5 | 1.9 | 0.15 | 72 | 35 |
| Ca-ZK-5 | 1.9 | 0.23 | 47 | 35 |
| SAPO-17 | 1.3 | 0.31 | 23 | 36 |
| SAPO-STA-7 | 1.7 | — | — | 37 |
| SAPO-34 | 1.6 | — | — | 38 |
| Na-SAPO-34 | 2.1 | — | — | 39 |
| Sr-SAPO-34 | 3.1 | — | — | 39 |
| SAPO-35 | 1.8 | 0.32 | 33 | 36 |
| SAPO-56 | 2.8 | 0.39 | 42 | 36 |
| SAPO-RHO | 1.2 | 0.086 | 84 | 36 |
| AlPO-17 | 0.66 | 0.14 | 25 | 40 |
| AlPO-18 | 0.52 | 0.13 | 22 | 40 |
| AlPO-25 | 0.21 | 0.068 | 18 | 40 |
| AlPO-53 | 0.90 | 0.031 | 170 | 40 |
The scope of this short review will be narrow and concise and focuses mainly on sorbents based on zeolite and related sorbents with narrow pore windows. The CO2 separation and sorption capability of these sorbents with narrow pore windows will be explored.
Adsorbents with narrow pore openings
Zeolites
Zeolites are porous and crystalline aluminosilicates that are both naturally occurring and can be synthesized. These covalent oxides of Al and Si form porous structures with interconnected channels or cages. The zeolite frameworks are negatively charged due to the difference in the oxidation states of Al(III) and Si(IV). The negative charges are balanced by exchangeable cations. Even though they share the common chemical formula of Mk+x/k[AlxSiyO2(x+y)] ZH2O (where M+ is the exchangeable cation), there are many different zeolite structures documented to date. These porous zeolites display a significant structural diversity with quite different pore sizes, pore openings, and topologies. The internal pore volume of zeolites is available for adsorption of small molecules and has been utilized in numerous industrial and household applications.The sorption properties of zeolites were very well studied by Barrer and co-workers in their early work.8–15 Their work focused mainly on natural zeolites, such as chabazite,8,10,11,16 mordenite,10,13,15 and analcite.17,18 Their studies included size or interaction based selectivities exhibited by zeolites on different sorbates. In addition, they were one of the first to study diffusion of different small organic and inorganic gas molecules in zeolites. We appreciate and acknowledge their work; however, they did not put significant focus on CO2 sorption. There is a vast amount of previous work on zeolites. Some properties of these materials, including their catalytic,19–21 ion exchange22–25 as well as gas separation/sorption1,26 properties. To make this review comprehensive, we focused on the literature related to CO2 sorption. In specific, a number of zeolite materials with narrow pore windows were considered.
CHA-Zeolite (chabazite)
The zeolite chabazite (CHA – shown in Fig. 1) is one of the most studied zeolites with narrow pore windows. It has a highly accessible porous framework of the 8-ring class with exchangeable cation sites. It exists naturally but can also be synthesized.41 As the cations can very easily be exchanged, many forms of zeolite chabazite exist. Barrer and associates studied the sorption properties of natural chabazite in detail.8,10–12,17,41 Zeolite chabazite can occlude and separate molecules by their size. This property first shown by Barrer and Ibbiston.17 Zeolite chabazite occluded small straight chain hydrocarbons but branched hydrocarbons were completely excluded. This separation ability was due to the narrow pore windows of chabazite (0.38 × 0.38 nm).42,43 More of zeolite chabazite's ability to separate different gas molecules was demonstrated further by Janák et al.44 and many of the work from Webley's group.27,45,46 Webley and associates observed that CO2 adsorbed significantly more on all their zeolite chabazite samples when compared with N2 and CH4.27 Zeolite chabazite in its K+ form (K-CHA) had enhanced ability to separate CO2 from N2 and CH4. The CO2 uptake of K-CHA, Na-CHA and K-CHA at 113 kPa (273 K) was around 5 times higher than the N2 uptake (Fig. 2). Furthermore, at low pressures (1.0 kPa), this ratio (CO2 adsorbed: N2 adsorbed, per cavity) reached over 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for K-CHA. They attributed this finding to the fact that CO2 molecules could penetrate into the windows at low pressures, but the larger N2 was essentially blocked by the big K+ cation.
:1 for K-CHA. They attributed this finding to the fact that CO2 molecules could penetrate into the windows at low pressures, but the larger N2 was essentially blocked by the big K+ cation.
 | ||
| Fig. 1 Structure representation of the CHA structure as in chabazite (and SAPO-34). The yellow lines represent Si (or Al) bonds to O, O atoms are represented by red lines. | ||
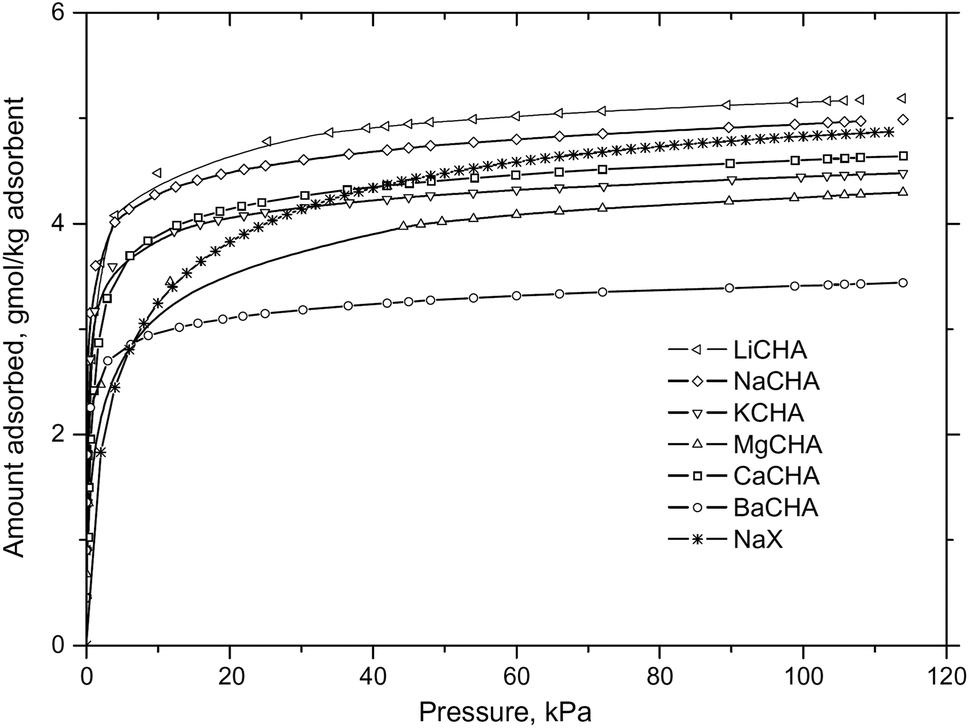 | ||
| Fig. 2 CO2 sorption isotherms of ion exchanged zeolite chabazite at 273 K, reproduced with permission.45 | ||
In terms of the capacity to adsorb CO2, previous literature shows that zeolites chabazite generally has a high capacity. Inui et al.47 showed that under pressure swing adsorption (PSA) conditions, zeolite chabazite had high uptake of CO2 (∼3.5 mmol g−1) and low irreversible uptake at high pressures (up to 1.1 MPa). Watson et al.48 demonstrated that the uptake of CO2 of a natural version of zeolite chabazite could reach over 5 mmol g−1 at a high pressure (3 MPa, 305 K). Na-CHA and Li-CHA both showed high uptake of CO2.45 The equilibrium uptake of CO2 at 120 kPa (273 K) was ∼4.4 mmol g−1 and 4.5 mmol g−1 for Na-CHA and Li-CHA, respectively. K-CHA, Mg-CHA and Ca-CHA showed CO2 uptake of ∼4.0 mmol g−1 under the same conditions.45 Ba-CHA showed a slightly lower uptake of CO2 (∼3.5 mmol g−1) under those conditions. The uptake of CO2 at low and close to zero loading was higher on Ba-CHA than on Li-CHA. The high uptake at low pressures may be related to the strong cation-quadrupole interaction for Ba2+ cation and CO2. This trend illustrates that the cation charge density, the electrical field gradients of the material and the interaction with the quadrupole moment of CO2 all are important. The original study (Zhang et al.45) gave detailed analysis into these observations. Zhang et al.45 also examined the CO2 isotherms of different ion exchanged chabazite materials in detail. They considered the dependence of the enthalpy of CO2 sorption on the cation. For Li-CHA and Na-CHA, the enthalpy of CO2 sorption increased with increased loading. Their findings agreed with the suggested explanations for the uptake dependencies on the cations, at different pressures. For Ba-CHA, Mg-CHA and K-CHA, the enthalpy of CO2 sorption dropped at high loading. This was rationalized and related to a decrease of the cation-quadrupole interaction, and that the sorbate–sorbate interaction in these materials was not dominant. In the case of Ca-CHA, the enthalpy of CO2 sorption stayed fairly constant with an increased loading, indicative of a balanced contribution from sorbate–sorbate interaction and cation-quadrupole interaction. These findings were corroborated by the high uptake of CO2 observed on Ba-CHA at very low pressures of CO2.
In short, many of Webley's and associates' work suggested that K-CHA can be a suitable CO2 sorbent of the zeolite chabazite family. K-CHA showed a higher preferential CO2 uptake over N2 than Li-CHA and Na-CHA, as well as a high CO2 capacity (although slightly lower than Li-CHA). They suggested that the enhanced CO2 selectivity was due to the large K+ ion close to the 8 MR window blocking N2 access into the pores.27 Many other ion exchanged chabazites also showed very good potentials to be CO2 sorbents. More recently, they proposed a “molecular trapdoor” mechanism to explain the enhanced selectivity of these ion exchanged chabazites.49,50 They explained that the very low uptake (essentially blocked) of bigger molecules such as N2 and CH4 was not entirely due to the size effect. Instead, they proposed that CO2 would interact with the cation strongly enough that the cation deviate from its “normal” site, allowing enough space for CO2 to enter the pores. Those that have weaker interaction with the cations (N2, CH4) do not interact and induce movement of the cation. They concluded that for this mechanism to work properly, the Si–Al ratio needs to be tuned. A low Si:Al ratio of around 1.5:1 is preferred to increase the CO2 selectivity. At this ratio, all “pore aperture doorways” are occupied by cations, which can restrict the adsorption of the N2 and CH4.50 This principle may also be applied to other small pore zeolites such as zeolite A.50
LTA-Zeolite A
Zeolite A has been studied extensively, similarly to chabazite. Zeolite A (LTA – Linde Type A) was first reported by Breck in 1956.51 It is a crystalline aluminosilicate with large cages and narrow pore openings (8-rings) with a number of charge balancing cations. In zeolite A, the Si–Al ratio is strictly 1:1, unlike in chabazite, which can have a higher Si–Al ratio.45 As a result of the large charge on the framework and the narrow pore openings, the electrical field gradient on zeolite A is typically very high.
Zeolite A (Fig. 3) has a cubic structure. The effective size of its windows are heavily dependent on the specific cation present. Monovalent cations tend to populate sites close to the 8-rings, while divalent cations tend to populate sites that do not partially block the 8-rings. Zeolite A with Na+ as cation has a pore window size of around 0.38 nm and is also called as zeolite 4A due to its pore windows of ∼0.38 nm in diameter. This pore window aperture can be adjusted to 0.5 nm or down to 0.3 nm, should the framework contain Ca2+ and K+ ions instead, respectively. Zeolite KA is also called zeolite 3A and zeolite CaA is also called zeolite 5A.
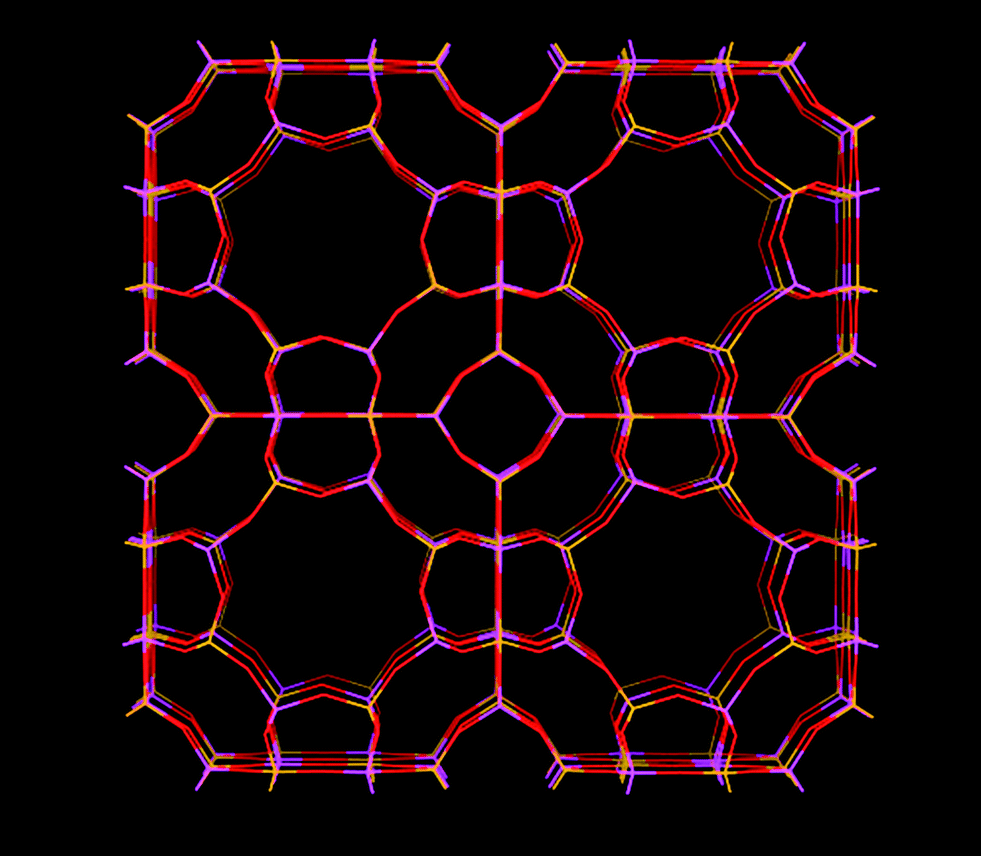 | ||
| Fig. 3 Structure representation of zeolite A, yellow lines represent Si bonds to O, pink lines represent Al bonds to O, O atoms are represented by red lines. | ||
The high electrical field gradients of zeolite A may also be responsible to its relatively high uptake of CO2. In an early study by Harper et al.,52 the capacity to adsorb CO2 on zeolite NaA was found to be ∼6.7 mmol g−1 at saturation. Those adsorption measurements were carried out at a temperature of 194 K (where CO2 saturation occurs at atmospheric pressure). At 273 K, they observed that the capacity to adsorb CO2 was still as high as 4.1 mmol g−1 (101 kPa). Bae et al.29 evaluated a range of different cation exchanged zeolite A for their CO2/N2 separation potential. They found that at the relevant pressure range, Ca2+ exchanged zeolite A (CaA) had an impressively high CO2 uptake (∼5.0 mmol g−1, 298 K) and a CO2/N2 selectivity of 250 (predicted by the authors using the ideal adsorption solution theory – IAST). They compared their results with Mg-MOF-74 and found that CaA had a higher volumetric uptake of CO2 (0.15 bar CO2, 313 K), higher working capacity (based on their TSA study) as well as a longer breakthrough time than MOF-74. Palomino et al.53 tested zeolite A with high Si:Al ratios (up to 5) and observed that the capacity to adsorb CO2 varied with the Si:Al ratio. They observed that the CO2 uptake at 500 kPa (303 K) was the highest for an intermediate Si:Al = 2:1. The CO2 uptake was lower on zeolite A with both lower and higher Si:Al ratios than for a ratio of 2. In addition, they observed that the isosteric heat of CO2 adsorption (up to 2.5 mmol g−1 loading) decreased with an increasing Si content. At high Si content, the regenerability of the zeolite A sorbent increased because of the lower heat of CO2 adsorption.53 The difference in the heat of adsorption is possibly due to the different number of cations in the zeolite, as CO2 tends to adsorb more strongly at high energy sites close to the cations (discussed in more details later). Palomino et al.53 also found that the heat of CH4 adsorption was not significantly affected by the difference in Si content, but the CO2/CH4 selectivity was reduced with increasing Si content. Inui et al.47 highlighted the high capacities to adsorb CO2 (3–4 mmol g−1, at 1.0–1.2 MPa) of zeolite NaA and CaA in an independent study. Due to the high electrical field gradients, the enthalpy of CO2 sorption on zeolite A is high. Bae et al.29 found that CaA had a noticeably higher heat of CO2 adsorption than NaA, and MgA. The heat of CO2 adsorption on CaA was around 60 kJ mol−1 at low loading, and decreased to around 30 kJ mol−1 with a loading of around 4 mmol g−1. They attributed the high heat of CO2 adsorption to the large number of accessible strong adsorption sites. Delaval and de Lara54 showed that CO2 physisorption on zeolite 4A had an enthalpy of around 50 kJ mol−1 at zero loading. The enthalpy change reduced with increased loading down to ∼44 kJ mol−1. We previously observed that the enthalpy of CO2 physisorption on zeolite NaKA was around 37 kJ mol−1 at nonzero loading.55 The low value we observed may be due to the presence of the big K+ cations.
As mentioned, the window size of zeolite A can essentially be further adjusted by ion exchange. We recently demonstrated that partially K+ ion exchanged zeolite NaKA had pore sizes between 0.3 and 0.4 nm.28 Using this feature of zeolite A, we were able to produce zeolite NaKA with 17% of the cations being K+, 83% being Na+. This zeolite, with the reduced pore size, was able to exclude N2 from sorption onto the material (<0.01 mmol g−1, 273 K, 101 kPa). The CO2-over-N2 relative uptake of the material reached over 200. The CO2 capacity of this highly selective zeolite NaKA remained high (3.5 mmol g−1, 273 K, 101 kPa). Mace et al.56 suggested that the high selectivity was not solely due to the bigger cation blocking the bigger sorbates. They concluded that the difference in mobility between Na+ and K+ and the higher interaction with CO2 allowed CO2 to enter the pores (when the material is not fully K+ exchanged). Other sorbates, such as N2, did not have the ability to do so.
The exclusion of N2 from sorption on zeolite NaKA appeared to be related to its large effective kinetic diameter (0.36 nm). Further reducing the pore window size of zeolite NaKA with additional K+ ions in the 8-ring, will make the apertures too narrow for CO2 to pass through, as the effective kinetic diameter of CO2 is about ∼0.33 nm. However, we observed significant capacities to adsorb CO2 also for zeolite NaKA with a high content of K+.28 Different mechanisms have since been proposed to rationalize this unexpected phenomenon. Larin et al.57 suggested that chemical reactions of CO2 with the framework atoms would lead to carbonate formation on the K+ cations near the 8-rings (as K2CO3 with one other K+ cation). They proposed that such carbonates would reposition the K+ atoms away from the window aperture. This would have resulted in a wider opening for CO2 to enter subsequently. Webley and associates,49,50 although did not study zeolite A explicitly, proposed “molecular trap door” mechanism for CO2 entering pores of chabazite when the material had been K+ exchanged. They stated in their conclusion that they expected to find a similar mechanism on zeolite LTA. The KCHA in their study also had pores that were theoretically blocked for CO2 to enter. As discussed earlier, they proposed that CO2 can interact and shift the position of the cation, allowing itself to enter the pores. Recently, Mace et al.58 presented a procedure using ab initio molecular dynamics calculations to access the details of the free energy barriers for diffusion of small gas molecules through 8-ring zeolite windows. By introducing certain spatial constraints, the gas molecule could be steered towards the “rare event” of the diffusion through the pore window of interest, without losing other relevant degrees of freedom. In this work, using this procedure, the free energy barriers of diffusion for CO2 and N2 in zeolite NaKA were estimated, investigating the differential molecular sieving effect of the two cation types, Na+ and K+, without involving either chemisorption or explicit “molecular trap door” mechanisms. The results were in good qualitative agreement with the experimental results presented by Liu et al.28 showing a drastic increase in the energy barrier for CO2 or N2 to pass a K+ blocked pore window compared to a Na+ blocked one, hence strongly supporting the idea of a tunable sieving effect through ion exchange.
The molecular details of CO2 sorption on zeolite A have also been studied. Jaramillo and Chandross59 studied CO2 (physi-)sorption using Gibbs ensemble Monte Carlo simulations. They suggested that CO2 sorption at low pressures occurred at a single cation Na+ site around the 6-ring windows. CO2 next adsorbed on a second site where it coordinated with Na+ from both the 6- and 8-rings. Finally, CO2 adsorbed on a third site where it coordinated to 3 Na+ cations (4-, 6- and 8-rings). Other evidence of CO2 adsorption on different sites depending on (CO2) pressure (or loading) was presented in a study by Delaval and de Lara,54 as well as our recent study on nano-sized zeolite A.55 These studies involved infrared spectroscopy and observed that the characteristic band for physisorbed CO2 (ν3 – asymmetric stretching vibration mode) occurred at a higher frequency (2352 cm−1) and downshifted to a lower frequency up on further loading of CO2. Density Functional Theory (DFT) studies showed that sorption of CO2 at a low coverage (1 CO2/α cage) occurred with CO2 bridging between 2 or 3 cation, irrespective of the size of the cation.55 Bae et al.29 determined using a neutron diffraction technique that at low loading of CO2, there were two adsorption sites on CaA. One of these two sites was located close to the 6-rings where CO2 could interact with two Ca2+ (site A), the other site (site B) was located in the center of the 8-rings (Fig. 4).
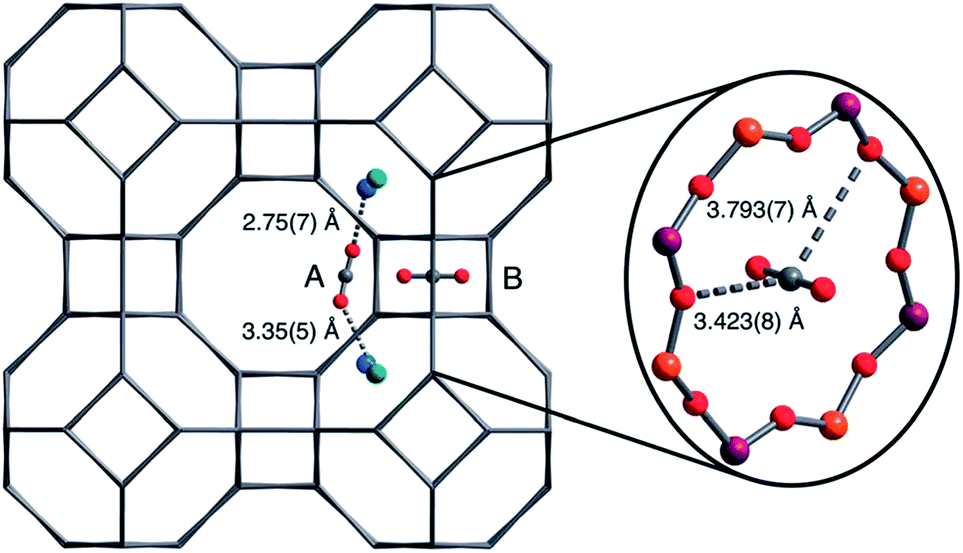 | ||
| Fig. 4 Location of the two CO2 adsorption sites in zeolite CaA at low loading, as determined using neutron diffraction by Bae et al.29 reproduced with permission. | ||
Many groups have studied diffusion of CO2 in zeolite A. This topic has recently been extensively reviewed by Ruthven.60,61 Here, we aim to give a short summary of previous work. Yucel and Ruthven62,63 concluded in their studies that CO2 diffusion in zeolite 4A was typically governed by intracrystalline diffusion (although diffusivity/mechanism was hugely dependent on the quality of the crystal). Surprisingly, they observed that zeolites of different origins can have quite different CO2 diffusivities. They attributed the differences to the subtle changes in the crystal structures and possibility due to the rearrangement of cations. The extent of dehydration could also be of importance, as shown by Kondis and Dranoff.64 In a recent publication by Ruthven,60 it was highlighted that the diffusion of sorbates in zeolite A is very complex. Many factors can significant alter the diffusivity of sorbates (such as CO2) in zeolite A. Zeolite crystals from different sources have very different sorbate diffusivity. The effect of different pre-treatment can also highly alter the diffusivity of sorbates such as CO2. We recently synthesized nano-sized zeolite A and studied the uptake rates of CO2 in this material. We found that the apparent diffusion was controlled by a skin layer on the surface of the crystals. We also observed zeolite A from different sources have very different sorbate diffusivity.55
RHO type zeolites
RHO type zeolites (Fig. 5) have shown interesting and desirable properties as a CO2 sorbent. RHO type zeolite is a synthetic zeolite with a cubic structure with narrow 8-ring pore openings. Typical Si:Al ratios are around 4 or 5:1. Different forms of this zeolite have shown high capacity to adsorb CO2. Palomino et al.32 showed that RHO type zeolite's (as-synthesized, containing Na+ and Cs+ cations) capacity to adsorb CO2 reached >6 mmol g−1 at a high pressure (∼850 kPa, 303 K). At atmospheric pressure (101 kPa, 303 K), its capacity to adsorb CO2 was still >3 mmol g−1. Araki et al.65 observed a similarly high capacity to adsorb CO2 on H+ exchanged RHO type zeolite (obtained by NH4+ exchange then calcination of as-synthesized RHO zeolite) that was synthesized using 18-crown-6 (18-C-6) as the organic structural directing agent (SDA). Its capacity to adsorb CO2 was ∼3.5 mmol g−1 (100 kPa, 298 K). The shape of the CO2 adsorption isotherm showed a step increase at low pressures. Lozinska et al.31 independently obtained cation free H-RHO. They showed that the cation free H-RHO adsorbed 3.3 mmol g−1 of CO2 at 80 kPa (298 K). The shape of the CO2 adsorption isotherm on their cation free H-RHO zeolite showed no step-wise increase as observed by Araki et al.65 The absence of this step-wise increase was due to the higher temperature used for the NH4+ exchanged used by Lozinska et al.31 which lead to a more complete ion exchange. The absence of Na+ and Cs+ in the cation free H-RHO zeolite obtained by Lozinska et al.31 meant that CO2 did not interact and move the cations (the effect of cation movement is discussed in the next paragraph).
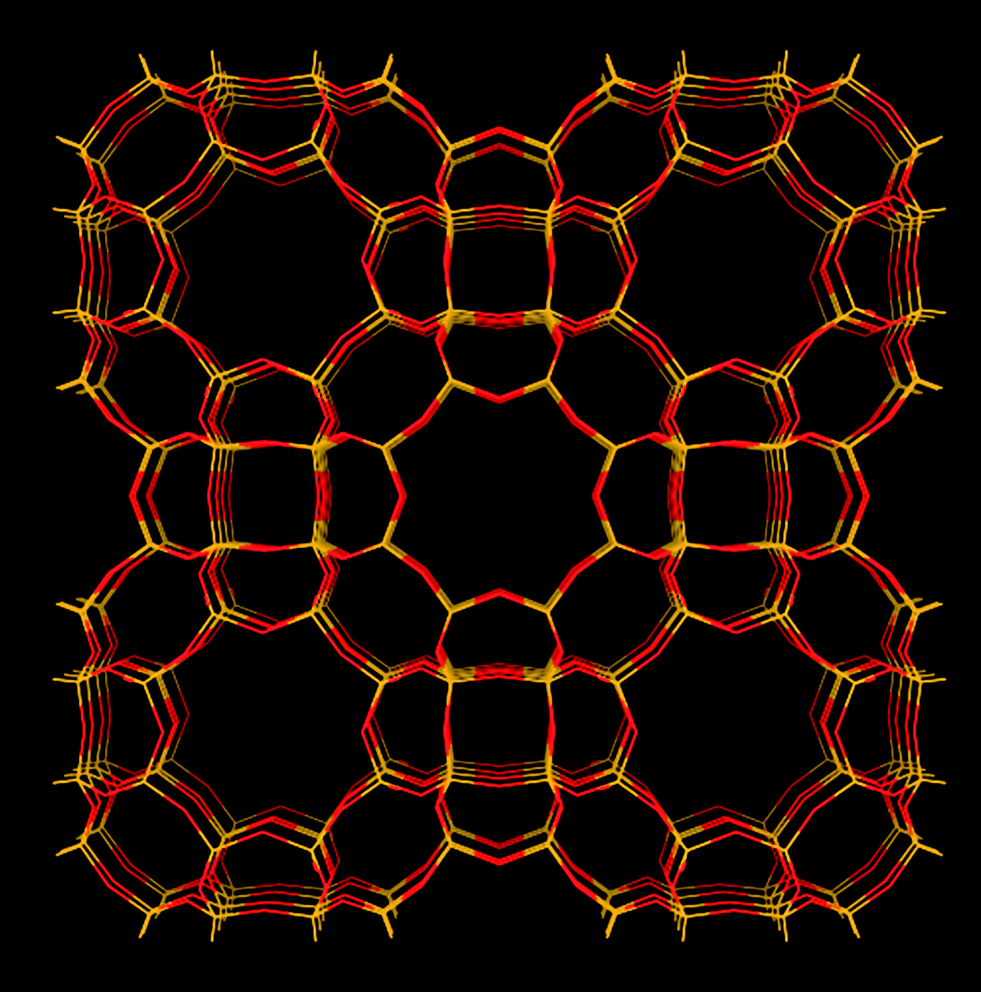 | ||
| Fig. 5 Structure representation of the RHO structure as in RHO type zeolite (and SAPO-RHO). The yellow lines represent Si (or Al) bonds to O, O atoms are represented by red lines. | ||
The high CO2 capacity of RHO type zeolite was in part due to its structure. Araki et al.65 observed that when the calcination temperature of as-synthesized RHO type zeolite was increased to >673 K, the CO2 capacity was reduced. They attributed this decreased CO2 uptake to the phase transformation of the RHO type zeolite at high temperature. At calcination temperatures >773 K, zeolite RHO synthesized with 18-C-6 was no longer stable. Palomino et al.32 argued that the uptake of CO2 caused the framework to expand and, hence, the capacity to adsorb CO2 increased as compared with a non-expanding framework. Such expansion was not observed for N2 or CH4, which both were restricted from entering the pores in the first place. Lozinska et al.31 showed that on cationic forms of RHO type zeolites (not H-RHO), CO2 interaction with the framework could “move” the cations (these cations otherwise block the pore window that would allow CO2 to enter) sufficiently to allow CO2 to enter and adsorb onto cationic RHO type zeolite. The repositioning of the cations changed the unit cell geometry (dimensions) of these zeolites and showed a step increase in the CO2 adsorptions. The step increase was not observed in the H-RHO zeolite of Lozinska et al.31 because of the absence of metal (large) cations. The high CO2 uptake was due to the high pore volume of this zeolite, and not caused by the CO2 induced repositioning of the cations. The framework effect for many kinds of RHO type zeolites with different cations was studied by Lozinska et al.,31 Lee et al.,66 Corbin et al.67,68 Nenoff et al.69 and Parise et al.70,71
RHO type zeolites appeared to have to a high CO2 selectivity over other gases such as CH4 or N2. Palomino et al.32 showed that the equilibrium selectivity (CO2-over-N2 relative uptake at 100 kPa) of zeolite RHO reached over 75. They attributed the high selectivity to the small pore diameter, as well as the high surface polarity of zeolite RHO.32 The high selectivity occurs because CO2 interacts and enables the cations in 8-ring positions to move out of these windows, enabling the CO2 molecules to enter the pores of RHO-type materials. Cation movement of this kind cannot be induced by other sorbates such as N2 and CH4, therefore, adsorption of N2 and CH4 on cation RHO zeolites appears as significantly hindered.
Other zeolites
Many other zeolites have also been investigated for their CO2 sorption and separation capability. Zeolite clinoptilolite also showed some interesting CO2 sorption properties. Barrer et al.10,72 and Inui et al.47 independently showed a high CO2 uptake on clinoptilolite at high pressures (3.7 mmol g−1 at saturation). Barrer and Murphy showed that the CO2 uptake would increase if the Si/Al ratio of clinoptilolite was increased (to 4.73 mmol g−1 at saturation when Si:Al reached 70:1).72 Aguilar-Armenta et al.73 showed that the CO2 adsorption kinetics on clinoptilolite was faster than other gases such as O2, N2 and CH4. All the mentioned studies stated that the enthalpy of CO2 sorption on clinoptilolite was very high (∼59 kJ mol−1 at zero loading)72 for all ion exchanged forms.73 Triebe and Tezel74 specifically mentioned that CO2 sorption on clinoptilolite was too strong for them to extract interpretable data from the gas chromatography study. The enthalpy of CO2 sorption was higher than O2, N2 and CH4 as well, giving clinoptilolite enhanced CO2 selectivity over these other gases. However, the high enthalpy of CO2 sorption meant that CO2 was difficult to remove from the material, as demonstrated by Inui et al.47 This would decrease clinoptilolite's appeal as a CO2 sorbent under cyclic sorption processes.
Other studies on zeolites with narrow pore opening as CO2 sorbents include zeolite T and ZK-5. Zeolite T is a narrow pore zeolite with a structure that is the result of the intergrowth of the erionite and offretite structures. The structural details of zeolite T had been clearly examined and discussed in literature.75,76 Jiang et al.34 and Cui et al.33 studied zeolite T membranes for CO2 separation from N2 and CH4. They found that the narrow pore openings of zeolite T (0.36 × 0.51 nm) could relate to the high selectivity observed. Both studies observed preferential CO2 uptake on these membranes. The equilibrium CO2 uptake was just above 3 mmol g−1 (298 K, atmospheric pressure, note that Cui et al. observed a higher uptake of CO2 ∼3.6 mmol g−1 under similar conditions).33,34 In mixed gas permeation experiments, Cui et al. found that the CO2/N2 and CO2/CH4 selectivity were 107 and 400, respectively.33 Zeolite ZK-5 adopts the KFI structure type. It is a high silica zeolite with the Si:Al ratio that varies from 4:1 to 5.1:1. Like other zeolites, it has ion exchange properties.77 Liu et al.35 studied zeolite ZK-5 (Fig. 6) and the ion exchanged forms of ZK-5. They found that H-ZK-5 had the highest CO2 uptake at 101 kPa (303 K) of 5.0 mmol g−1. On the other hand, they showed that Mg-ZK-5 had high working capacity in the pressure region related to PSA application. Furthermore, Li+, Na+, and K+ exchanged ZK-5 showed better working capacity for CO2 and higher CO2 selectivity over N2 in the pressure regions relevant for vacuum swing adsorption (VSA). Remy et al.78 synthesized a low silica version of zeolite ZK-5 (LS-KFI) with Si:Al ratio of around 1.6:1. They found that LS-Li- and LS-Na-KFI had higher CO2 capacity than zeolite Li- and Na-ZK-5 (Si:Al = 3.6:1) at low pressures. The higher CO2 adsorption at low pressures was due to the increased electrostatic interaction between CO2 and the cations. They also found that zeolite ZK-5 with higher Si content than LS-KFI have higher CO2 working capacity (in CO2/CH4 separation). LS-KFI was more selective for CO2 over CH4 than zeolite ZK-5, due to the higher number of cations in LS-KFI than ZK-5. They did not study CO2 separation from N2, but similar responses in the CO2 over N2 selectivity could be expected on LS-KFI and zeolite that ZK-5.78
 | ||
| Fig. 6 Structure representation of the KFI structure as in zeolite ZK-5. The yellow lines represent Si (or Al) bonds to O, O atoms are represented by red lines. | ||
Silicoaluminophosphates and aluminophosphates
Certain crystalline and porous phosphates, in particular silicoaluminophosphates (SAPOs) and aluminosphophates (AlPOs), can have narrow pore openings with 8-ring windows. These classes of phosphates were first synthesized in the early 1980s.79–81 The structure of AlPO is somewhat similar to microporous silicas.40 AlPOs are made from covalent oxides of Al and P connected together. Phosphorus has an oxidation state of (V) in the AlPO. This results in a neutral framework with no charge balancing cations, as for microporous silicas. SAPOs on the other hand are similar to zeolites as they have negatively charged frameworks. The framework structures of SAPO are composed of oxides of Al, Si and P, and the crystallization of SAPOs appears to proceed via an AlPO intermediate.82,83 After the formation of such an AlPO intermediate, Si(IV) replaces P(V) in the framework, creating SAPOs. The P replacement by Si creates negative charges on the SAPO framework due to the lower oxidation state of Si(IV). As for zeolites, these charges are balanced by exchangeable cations.The difference between a neutral and negatively charged framework can be significant. The neutral framework on AlPOs means that the material has a much lower overall electrical field gradient than on SAPOs. These gradients are not as low as those on microporous silicas, due to the more ionic character of the oxides in AlPOs.84 Consequently, unlike for zeolites, we showed that AlPOs displayed somewhat hydrophobic properties.40 As expected, the negatively charged framework on SAPOs gives the materials higher electrical field gradients, but not as high as low silica zeolites (e.g. zeolite A). The field gradients also make them more hydrophilic than AlPOs, but less than zeolites.85 In applications where the gas streams contain a significant partial pressure of water, the difference in hydrophilicity can be of significance.
Both SAPOs and AlPOs can adopt structures that are analogues of zeolites. One of the most studied phosphates, SAPO-34, has the same overall structure as zeolite chabazite (CHA).86 This compound is easily synthesized and has been in the focus of many studies related to catalysis, ion exchange and gas sorption. It is highly porous with active cation sites. Other phosphates structures, such as AlPO-5 and SAPO-5 (AFI), have no zeolite analogues (although the pure silica analogue exists). AlPO-5 and SAPO-5 are also widely studied due to their large 12-ring pore channel with a very smooth surface.87–89 These materials are seen to be potential catalysts in some applications because of high diffusion rate of guest molecules into the pore channel system.
Below different SAPO and AlPO materials with narrow pore openings are reviewed. The previous findings on their CO2 sorption and separation properties are summarized.
Silicoaluminophosphates with narrow pore openings
A number of SAPO materials with narrow pore openings have been studied for numerous applications. The most notable example is SAPO-34 (CHA).38,39,90–93SAPO-34 (structure shown in Fig. 1) is commonly studied not only because of its properties, but also due to the easy synthesis and the high purity of the synthetic product. It has been found to be stable under humid atmosphere at temperatures >373 K, although care must be taken as the adsorption of water generally affect the long term stability of this material.93 The chabazite framework (Fig. 1) also allows for fast diffusion of small gas molecules due to its windows' dimensions (0.38 × 0.38 nm). Many studies have examined the high CO2 capacity of SAPO-34.39,92,94 The CO2 uptake on SAPO-34 reached over 3.5 mmol g−1 at 295 K (101 kPa).95
Ion exchanged SAPOs have been studied extensively. One important note on ion exchanged SAPOs is that, unless carefully performed, SAPOs tend to lose their crystallinity upon ion exchange. A possibly reason for the lost in crystallinity is the high concentration of H+ that is released during ion exchange, which can destroy the SAPO framework. The focus on ion exchanged SAPOs has somewhat been on SAPO-34, particularly Sr-SAPO-34. Arévalo-Hidalgo et al.,92 Hong et al.38 and Rivera-Ramos et al.39 are just some of many studies that have found that Sr-SAPO-34 had the best overall adsorption performance for CO2 of the different SAPO-34 variations. They found that the adsorption capacity for CO2 was enhanced by the Sr2+ cation, especially at low partial pressures of CO2. Rivera-Ramos et al.39 argued that Sr-SAPO-34 performed better than the Ce3+, Ti3+, Mg2+, Ca2+, Ag+ or Na+ exchanged SAPO-34. They suggested that the Sr2+ cations are easily accessible but without causing any transport resistance or pore blocking. The low stability of Ce3+ and Ti3+ (and Ti4+) cations for ion exchange on SAPOs/zeolites needs to be considered. On the other hand, they assumed that Ce3+ and Ti3+ cations blocked the pores by occupying the S'III site, giving these variants of SAPO-34 very low CO2 uptakes. Arévalo-Hidalgo et al.92 had similar observations for Na-SAPO-34 and Ba-SAPO-34. They too suggested that the cation sites for Sr2+ (and Ba2+) are located in a position where CO2 interaction will consequently become strong.
Takeguchi et al.94 incorporated Cu2+, Fe3+, Ni2+ into the SAPO-34 (CHA) framework to concentrate and separate CO2 from N2-diluted gaseous mixture in a PSA apparatus. They observed that SAPO-34 and, in particular, Ni-SAPO-34 have high CO2 separation and uptake capacities. Ni-SAPO-34 was able to concentrate CO2 from a gas stream with a CO2 concentration of 2.9% up to 84.4% with a high CO2 recovery up to 33%. They compared these Ni incorporated SAPO-34 with zeolite ZSM-34 (silica version of zeolite T, mixed ERI and OFF phases) and SAPO-20 (a 6-ring material). They concluded that metal incorporated SAPO-34 had the best properties for CO2 separation from a mixture with N2, when compared with the other materials they studied.
Several groups synthesized SAPO-34 onto membranes for gas separation testing. Different gas pairs were investigated for such inorganic membranes of SAPO-34 including CO2–CH4, H2–CH4, CO2–N2, N2–CH4 and other light gas mixtures.38,96–99 For non-ion exchanged SAPO-34 membrane, the difference in diffusivities of different gases allowed the membranes to show high selectivity towards CO2 over N2 and CH4.95
Separate from ion exchange, Venna and Carreon91 functionalized SAPO-34 on a membrane with amines (ethylenediamine, hexylamine and octylamine). They tested the properties of these amine impregnated SAPO-34 membranes with respect to their properties related to separation of CO2. Amine functionalization on such small pore materials has not been studied extensively. They observed with low amine loading, there was an improvement on the CO2 uptake due to CO2 interaction with amine groups. At high amine (ethylenediamine) loading, the capacity to CO2 adsorption and transport were adversely affected. The functionalized material, with low amine loading, showed steeper CO2 isotherms at low pressures and a higher equilibrium uptake overall.
Other SAPO materials with narrow pore openings have not been studied as much as SAPO-34, but some of the studied SAPOs display interesting CO2 separation properties. SAPO-STA-7 (SAV, Fig. 7), a material first synthesized by Castro at al.,37 showed very high CO2 uptake at high pressures. They studied the molecular and thermodynamic details of CO2 sorption. In contrast to AlPO-18 (discussed later), CO2 has two different adsorption sites on SAPO-STA-7. The heat of CO2 sorption was found to decrease with increased loading (from ∼38 to ∼25 kJ mol−1). They attributed this large change in heat to that the adsorption sites associated with a high heat of CO2 sorption were occupied first. When these sites were fully occupied, CO2 began to adsorb on less energetically favorable sites. We drew very similar conclusions for SAPO-35 (Fig. 8) and SAPO-56 (Fig. 9) using results from in situ IR spectroscopy.36 In that study, we found that CO2 sorption first occurred on high energy (strong) Lewis acid sites, where actually CO2 acted as a Lewis base. Su et al.100 studied SAPO-RHO (referred DNL-6, Fig. 5) and found that CO2 uptake was enhanced by the number of acid sites. They found that SAPO-RHO had the highest CO2 uptake at a medium level of Si incorporation (Si:Al 0.37:1). This level of Si incorporation corresponded to the highest concentration of acid sites. In general, CO2 sorption took place on the lower energy sites when the loading increased. This trend was clearly visible from the in situ IR spectra of CO2 sorption on both SAPO-35 and SAPO-56. The asymmetric stretching vibration mode of adsorbed CO2 downshifted from 2357 cm−1 to 2345 cm−1 with increased CO2 loading (frequencies for SAPO-56). In the same study, we studied a range of SAPO materials with narrow pore openings. SAPO-56 had very high CO2 capacity at 5.5 mmol g−1 at 273 K (101 kPa), this level of uptake was very comparable to the commercially available zeolite 13X sorbent. Other SAPOs, including SAPO-17 (ERI, Fig. 10), SAPO-35 and SAPO-RHO, all had respectable levels of equilibrium CO2 uptake at 237 K, 101 kPa (3.3 mmol g−1, 3.6 mmol g−1 and 3.6 mmol g−1, respectively). As expected with SAPOs with their lower electrical field gradients (than zeolites'), the shapes of the CO2 isotherms were less steep at low pressures. These less steep isotherms were partly due to the lower amount of chemisorbed CO2 on SAPOs than on zeolites such as zeolite NaA. Still, the equilibrium CO2/N2 selectivities of these SAPOs were not low. These phosphates were also found to be less hydrophilic than zeolite 13X, as shown by the shape of the water adsorption isotherm at low relative pressures. This tendency means that under slightly moist conditions, SAPO material will be less sensitive to the presence of water than typical zeolites.36 Hydrophilicity can be an important property of a CO2 sorbent, as the use of a non-water sensitive sorbent will significantly reduce the cost of drying the gas stream.
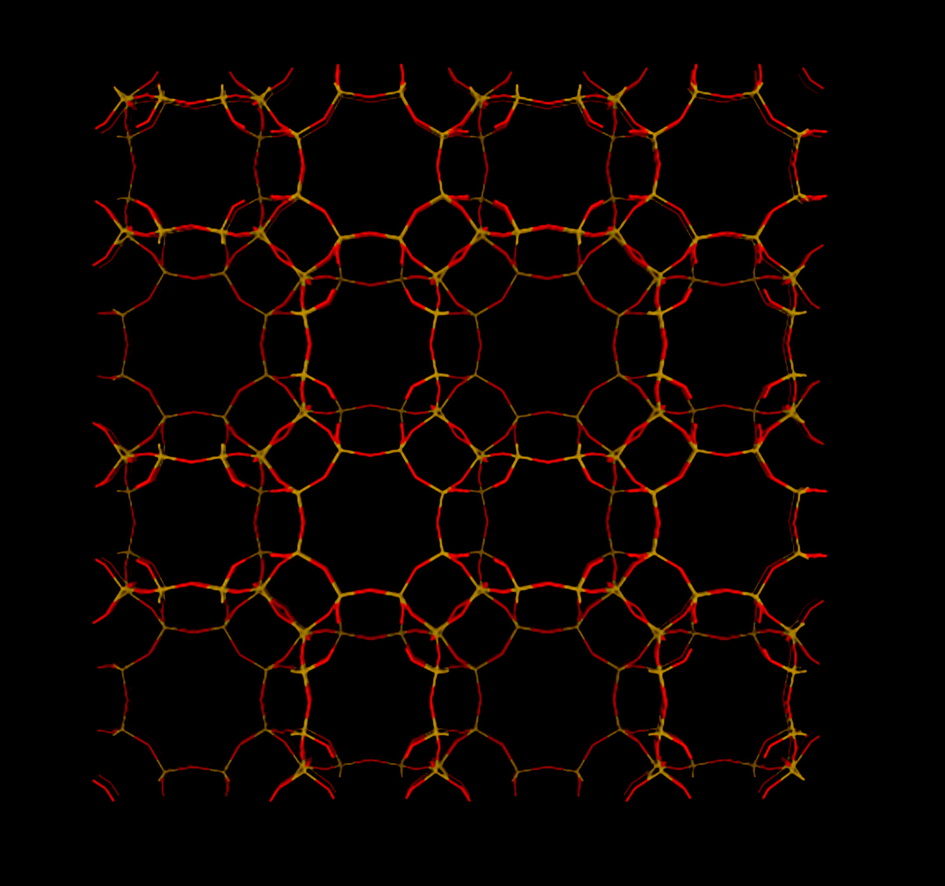 | ||
| Fig. 7 Structure representation of SAPO-STA-7 (SAV) The yellow lines represent Si (P or Al) bonds to O, O atoms are represented by red lines. | ||
 | ||
| Fig. 8 Structure representation of SAPO-35 (LEV), the yellow lines represent covalent bonds between two metal (Si, Al or P) atoms (bridging O atoms), oxygen atoms are purposely omitted in order to show the pore system more clearly. | ||
 | ||
| Fig. 9 Structure representation of SAPO-56 (AFX), the yellow lines represent covalent bonds between two metal (Si, Al or P) atoms (bridging O atoms), oxygen atoms are purposely omitted in order to show the pore system more clearly. | ||
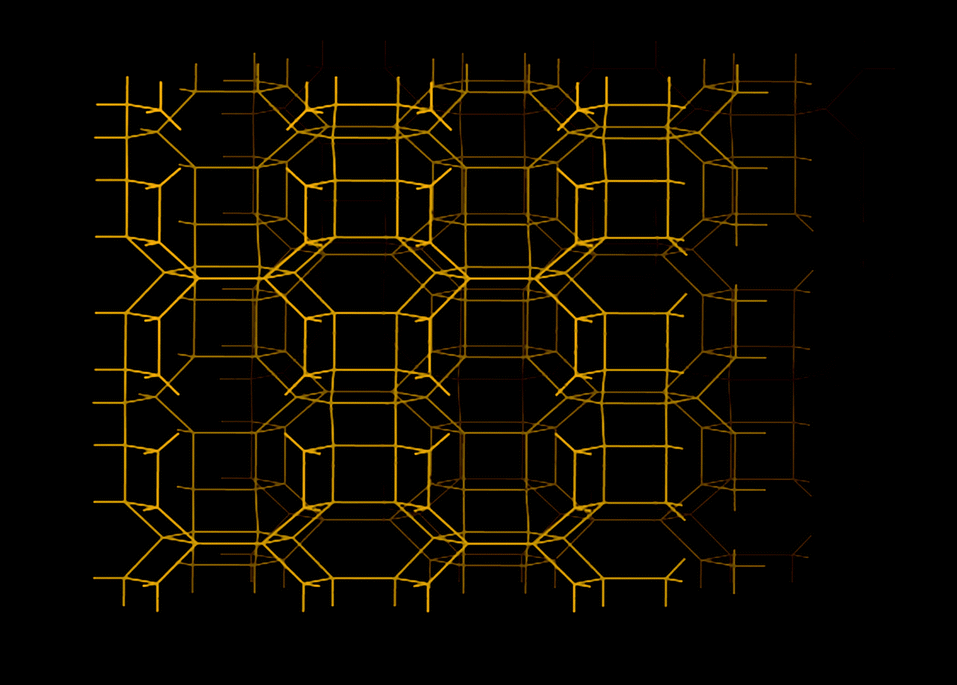 | ||
| Fig. 10 Structure representation of SAPO-17/AlPO-17 (ERI), the yellow lines represent covalent bonds between two metal (Si, Al or P) atoms (bridging O atoms), oxygen atoms are purposely omitted in order to show the pore system more clearly. | ||
Aluminophosphates with narrow pore openings
The low hydrophilicity is perhaps the most predominant feature of AlPO materials. These materials, with the lack of framework negative charges and charge balancing cations, have very low electrical field gradients. On the other hand, the electrical field gradients of AlPOs are still higher than microporous silica. The lower field gradients reduce the interaction between the material and water, making AlPOs somewhat hydrophobic. The water adsorption isotherms showed very little water uptake at low relative pressures, particularly for AlPO-53, Fig. 11.40 These properties was observed by us in our study related to a range of AlPOs with narrow pore openings, including AlPO-17 (ERI- Fig. 10), AlPO-18 (AEI), AlPO-21 (AWO), AlPO-25 (ATV) and AlPO-53 (AEN). The structures of these AlPOs are shown in Fig. 12.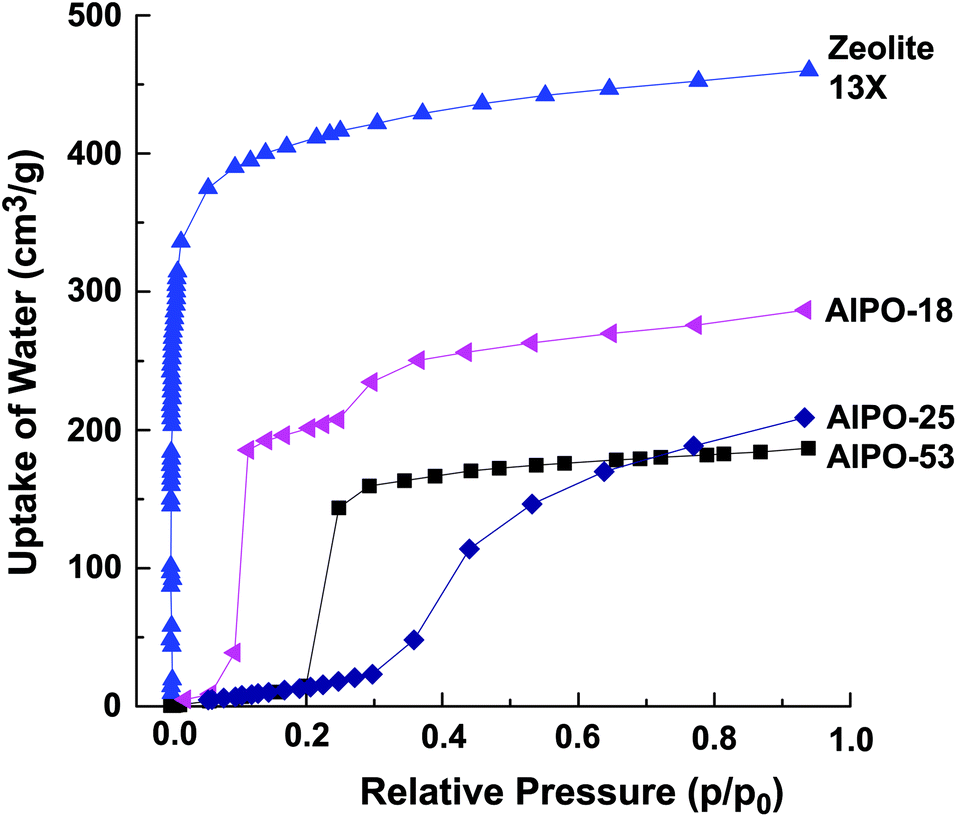 | ||
| Fig. 11 Water adsorption isotherms of various narrow pore AlPOs at 293 K, compared with zeolite 13X. | ||
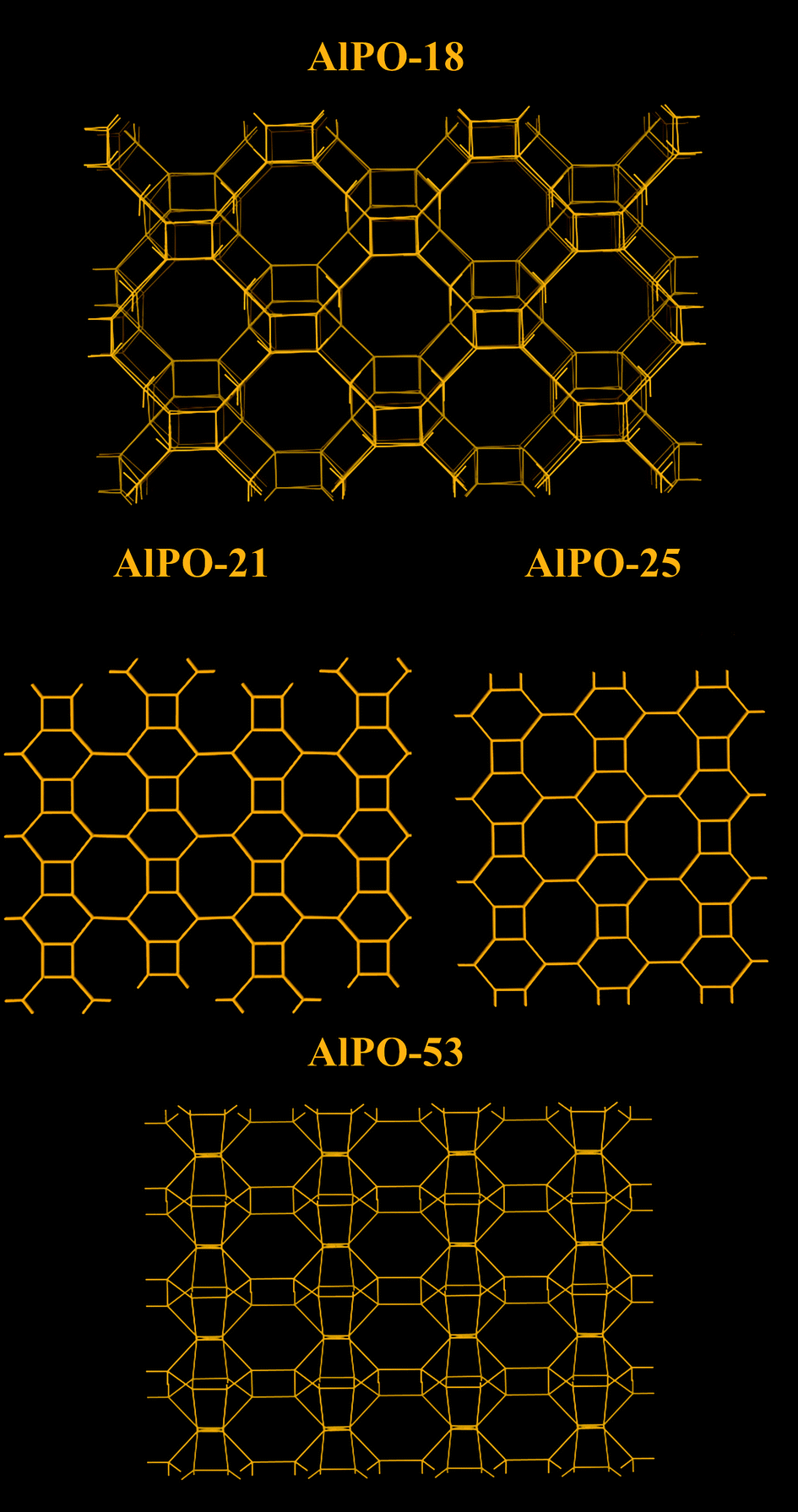 | ||
| Fig. 12 Structure representation of AlPO-18 (AEI), AlPO-21 (AWO), AlPO-25 (ATV) and AlPO-53 (AEN), the yellow lines represent covalent bonds between two metal (Al or P) atoms (bridging O atoms), oxygen atoms are purposely omitted in order to show the pore system more clearly. Note that AlPO-21 transforms to AlPO-25 upon calcination. | ||
The effects of the low electrical field gradients of AlPOs were not limited to the water uptake. AlPO-17, which has the same basic structure as SAPO-17, exhibited a lower equilibrium uptake of CO2 (at 101 kPa and 273 K).36,40 The uptake of CO2 of AlPO-17 was 2.3 mmol g−1 under these conditions (as compared to 3.3 mmol g−1 for SAPO-17). The lower uptake was in part, related to the lack of chemisorbed CO2 because of the absence cation sites. The shape of the CO2 isotherm of AlPO-17 had a more linear respond than its SAPO counterpart.
The equilibrium uptakes of CO2 of the other AlPOs were still significant, although not as high as for SAPOs or zeolites. This difference could be the reason why AlPOs are not as well studied as CO2 sorbents. The main explanation to the lower uptake of CO2 on AlPOs at the studied conditions is the weaker interaction between their frameworks and CO2. Nevertheless, some groups have studied AlPO-18 as a CO2 sorbent. Carreon et al.101 synthesized a AlPO-18 membrane for CO2 separation. The AlPO-18 membrane offered high selectivities for CO2/N2 (19) and CO2/CH4 (up to 59) with a high CO2 permeance of ∼6.4 × 10−8 mol s Pa m−2 (295 K). Wright and co-workers102 studied AlPO-18 both using computational and experimental approaches. They compared the shapes of the CO2 isotherms for AlPO-18 and a SAPO material (STA-7). The isotherms showed a much gentler slope on AlPO-18 in the low pressure region. This shape strongly suggested that AlPOs had lower enthalpy of sorption for CO2 than SAPOs. CO2 essentially experiences AlPOs as materials with relatively homogeneous surfaces. On AlPOs, there are no high energy adsorption sites, as one would expect to find on zeolites or even SAPOs. This absence was indicated by the increasing trend for the enthalpy of sorption with increased loading of CO2. Sorption did not first occur on any particular sites, and, hence, the enthalpy change during sorption was very similar or all sites. The increased enthalpy of sorption at high loadings was related to CO2 interacting with other already adsorbed molecules of CO2.
Interestingly, AlPOs have the ability to retain almost all of its capacity to adsorption of CO2 under cyclic adsorption conditions.40 This retained capacity has been related to the lack of chemisorption and high energy physisorption sites. After 5 adsorption cycles, AlPO-53 and AlPO-17 retained >99% of their original capacity. This would suggest that these materials would have a longer life time over many cycles. Furthermore, due to the more linear shape of the CO2 adsorption isotherms of AlPOs as compared with SAPOs and zeolites, AlPOs could be comparably more suitable for certain PSA based separations. The removal of CO2 from AlPOs during desorption can be effective and lead to a high working capacity in some industrial applications.
Microporous silicas with narrow pore openings
In the previous section, we considered the low electrical field gradients on AlPOs had both advantages and disadvantages in terms of their gas (and water) sorption properties. Microporous silicas belong to a related class of materials with low electrical field gradients. These silicas are essentially three dimensional covalent and crystalline SiO2 with internal pores. Hence, the framework of microporous silicas contain Si and O atoms only. The frameworks are neutral with no charge balancing cations. When compared with AlPOs, the electrical field gradients of microporous silicas are even lower, as the surface of the material is much more homogenous than AlPOs (with its two different electropositive atoms).28Because of the low electrical field gradients, the porous silica do not interact strongly with sorbates via the quadrupole-“electrical field gradient” mechanism. Despite of that, the capacity of CO2 sorption on certain microporous silicas can still be rather significant. Maghsoudi et al.103 studied Si-CHA and found that the uptake of CO2 reached ∼2 mmol g−1 (at 298 K and atmospheric pressures). Himeno et al.104 found that at higher pressure (3 MPa, 298 K), the uptake of CO2 on Si-CHA reached around 2.7 mmol g−1. The uptake was less than that of H2S but significantly higher than those of CH4 and N2 at all studied pressures.103 Maghsoudi et al. observed that its uptake of CO2 was 4.1 times higher than the uptake of CH4 at 100 kPa and 298 K.104 Miyamoto et al.105 observed that its uptake of CO2 was 19 times higher than its uptake of N2 at 75 kPa and 313 K and 5 times greater at high pressures (800 kPa, 313 K). With a CO2–N2 (equimolar) mixed gas, the uptake of N2 of the Si-CHA membrane became negligible even at high pressures (1.2 MPa).105 Similar results were observed on Si-DDR. Himeno et al.104 and van den Bergh et al.106,107 showed in their studies that the uptake of CO2 of membranes of Si-DDR membrane was 2–3 mmol g−1 at 273 K (120 kPa) and very high CO2 selectivity over N2 and CH4 (slight variations between the different studies, probably due to the different membranes). Separate permanence studies have shown that the CO2 selectivity over N2 and CH4 can reach 3000 (or around 40 for a CO2, N2 and CH4 mixture).106,107
The high CO2 selectivity observed was related to the sorbent–sorbate interaction. For a non-polar material with as low electrical field gradient as microporous silicas, the adsorption of sorbate is mainly based on dispersion and repulsion interactions. CO2 with its significant quadrupole moment is more easily polarizable than N2 and CH4. The quadrupole on CO2 can induce polarity on the SiO2 framework, increasing the sorbent–sorbate interaction.103 Another reason for this high selectivity is due to the window size of the materials, in particular Si-DDR. van den Bergh et al. concluded that the high selectivity was due to 3 different factors; steric effect (and consequently a kinetic effect) introduced by the small window opening of Si-DDR, competitive adsorption effect and the interaction between sorbent and sorbate, the latter two are very much enhanced for sorption of CO2.107
Due to the comparably low interaction between CO2 and the crystalline SiO2 framework, the enthalpy of CO2 sorption on these materials is low. In addition, these sorbents do not chemisorb CO2. Maghsoudi et al.103 found that enthalpy of CO2 sorption on Si-CHA was 21 kJ mol−1 (non-zero loading), which was significantly lower than on zeolites and phosphates. Himeno et al. established that enthalpy of CO2 sorption on Si-DDR was even lower (18.2 kJ mol−1 at nonzero loading), lower than some other crystalline and microporous silicas (∼32 kJ mol−1).104,108
Titanium silicates (titanosilicates) – ETS-4
Titanium silicate ETS-4 is built from covalently linked oxides of Ti and Si. It is a structure analogue of the minerial zorite,109 with a 3 dimensional framework. It has a 12-ring channels running along the crystallographic z axis and 8-ring channels running along the y axis (Fig. 13). Although not covered by this review, the ion exchange110,111 and catalytic112 properties of ETS-4s were found to be impressive. The as synthesized form of ETS-4, usually Na-ETS-4, becomes unstable when dehydrated. Some cation exchanged forms (mainly with divalent cations) of ETS-4 are more stable, as demonstrated by Anderson and Kuznicki et al.109,113 At high degrees of dehydration, ETS-4 often transforms into a related phase called as CTS-1 (contracted titanosilicate-1).113,114 Nevertheless, the pore size of CTS-1 can be controlled by dehydration under which it contracts. The contraction is not easily reversible. Nair et al. showed that dehydration of Sr-ETS-4 can “continuously vary(ing) the effective pore dimension”. Kuznicki et al., showed that Sr2+ exchanged ETS-4 can be used for separation CO2 from CH4, the material has since be put into application and carries a name of “Molecule Gate”.113–115 As shown separately by Park et al.116 the CO2 uptake of Sr-ETS-4 and other forms of ETS-4 varied significantly depending on the dehydration temperature. In their study, Ca-ETS-4 dehydrated at 373 K for 8 hours showed the highest uptake of ∼2.2 mmol g−1 (101 kPa, 298 K) of Ca, Sr and Ba-ETS-4 (Fig. 14). We are currently studying a range of ETS-4s in detail, paying particular attention to the transformation from ETS-4 and CTS-1. Anson et al.117 innovatively incorporated halogen atoms onto the framework of some ETS-4s during the synthesis step. The large halogen atoms were placed around the 8-ring windows and increased the CO2 over CH4 selectivity.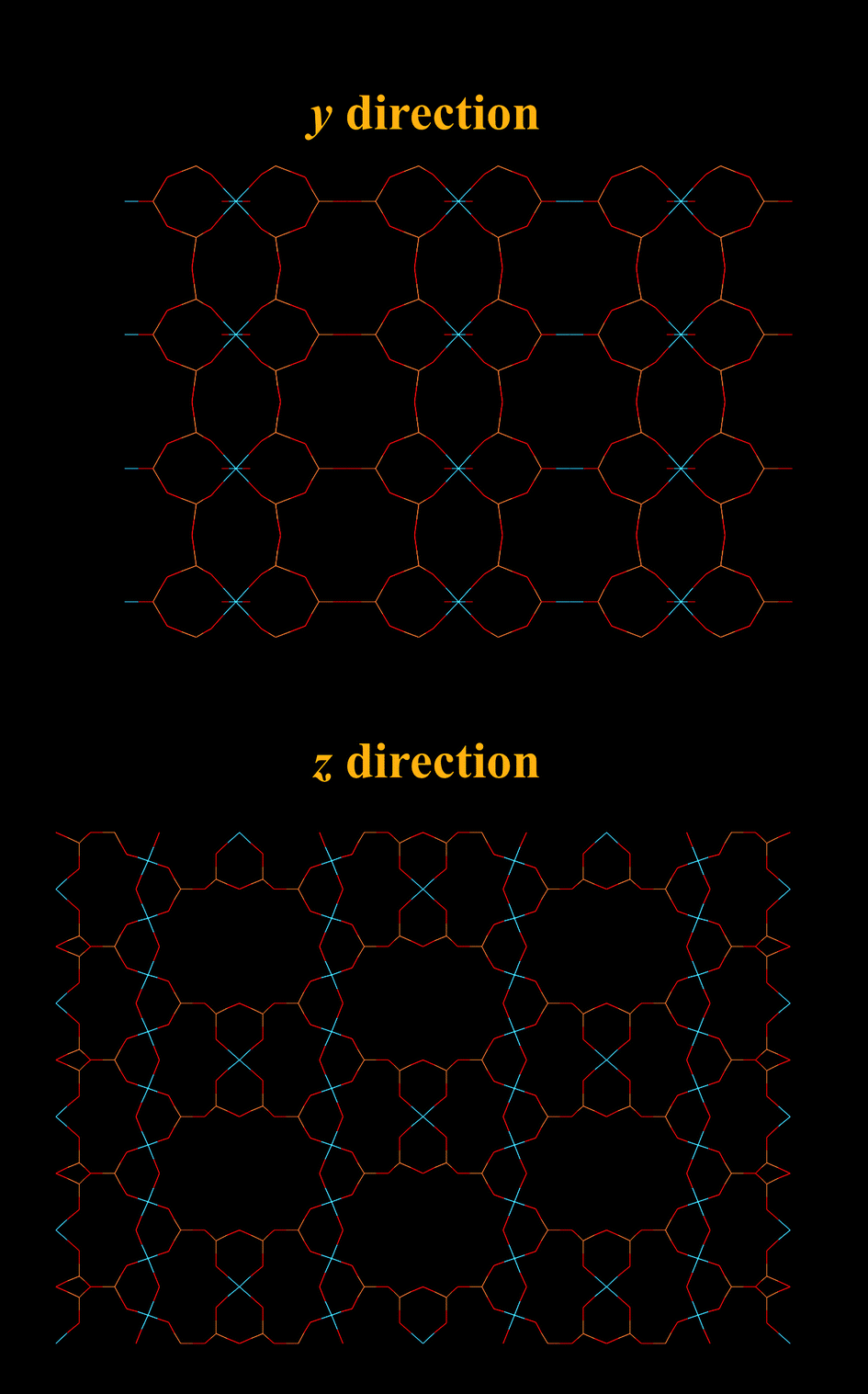 | ||
| Fig. 13 Structural representation of ETS-4, The yellow lines represent Si bonds to O, light blue lines represent Ti bonds to O, O atoms are represented by red lines. CTS-1 is a disordered, contracted version of ETS-4, the basic structure of the two appeared to be the same. | ||
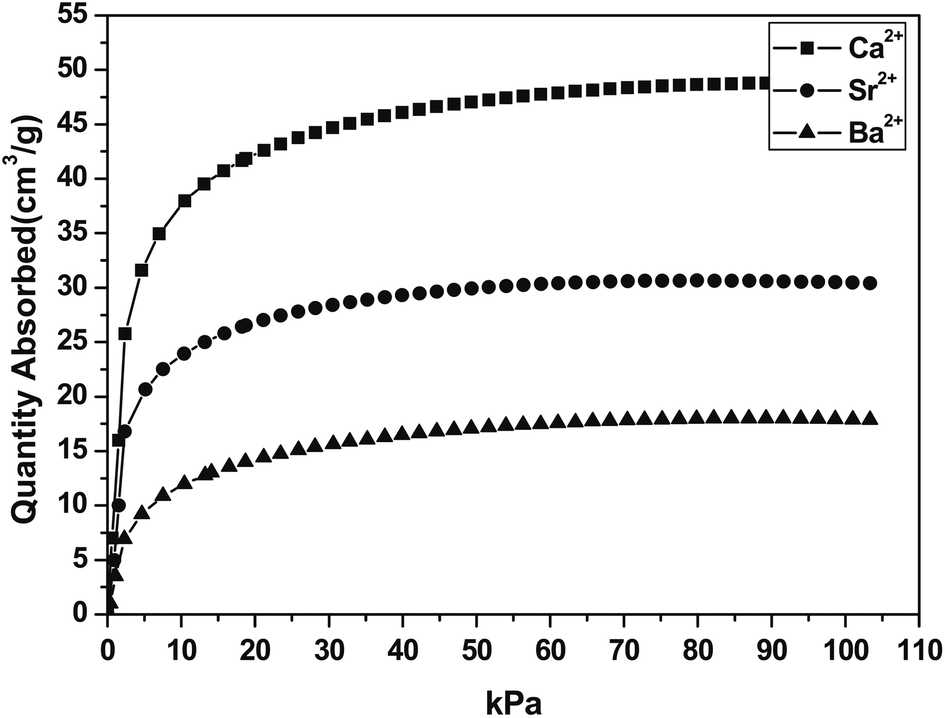 | ||
| Fig. 14 CO2 adsorption isotherms of Ca, Sr and Ba-ETS-4 at 298 K, samples prepared at 373 K. Reproduced with permission.116 | ||
Conclusion and outlook
A wide range of potential sorbents is available for the purpose of CO2 capture. Here, we explored some of the most studied inorganic porous sorbents with narrow pore openings. Each of the sorbents discussed here, of course, has its own advantages and drawbacks. Zeolites have been very thoroughly studied up to now and continuous efforts are being made. A main advantage of zeolites is the cost of manufacture. Both zeolite chabazite and zeolite A are commercially available and the properties of these materials can be easily tuned. Studies have found that zeolites offer high uptake of CO2 and certain variations have very high selectivity. The high electrical field gradients of zeolites are partly responsible for these features. Unfortunately, zeolites can adsorb CO2 very strongly, reducing the ease for their use in cyclic processes. Furthermore, zeolites are also hydrophilic. All silica zeolites (microporous silicas) can overcome both these problems. The low electrical field gradient weakens the strength of CO2 sorption and makes such silicas hydrophobic, yet still offering very high selectivity. As a result, microporous silicas can have good cyclic capacity for CO2. The comparatively low uptake of CO2 and somewhat tedious synthesis, as well as the high cost of mass manufacture are the major drawbacks of silicas. Should more efforts be put into developing zeolite based sorbents, the focus should be on simplifying and reducing the cost of producing microporous silicas.A middle way between zeolites and microporous silicas would direct towards the phoshpates materials. SAPOs offer equally high capacity for adsorption of CO2 as zeolites at relevant pressures. Their weaker electrical field gradients and lower number of cations result in highly reversible uptake of CO2 and lower sensitivity towards water. AlPOs are somewhat similar to SAPOs, with even lower sensitivity towards water, but the uptake of CO2 is also noticeably reduced at the relevant temperatures and pressures. They offer an impressive cyclic capacity; over 99% of the capacity to adsorption of CO2 was retained by AlPO-53 and AlPO-17 after 5 adsorption cycles. Even though AlPOs can be costly to synthesize, the potentially long lifetime may be an argument to develop these materials further.
A titanium silicate (ETS-4) has also been well studied as a CO2 sorbent and is already used in application for biogas upgrading. The tuneable pore size (by dehydration) is an attractive feature of using this material in application. ETS-4 has many potentials, further development of ETS-4 could provide very valuable outcome.
Taken all together, many of these narrow pore adsorbents have shown potentials for applications in CO2 separation, but several problems are yet to be overcome. We believe that intensified collaborations, between engineering groups and chemistry/physics groups would be especially beneficial for the further development of these sorbents.
Acknowledgements
The authors acknowledge the Swedish Energy Agency, and the Swedish Research Council (VR) and the Swedish Governmental Agency for Innovation Systems (VINNOVA) through the Berzelii Center EXSELENT for the respective research funding. Dr Jie Su is acknowledged for her help with Fig. 13. Amber Mace is acknowledged for her helpful input.Notes and references
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- M. M. F. Hasan, E. L. First and C. A. Floudas, Phys. Chem. Chem. Phys., 2013, 15, 17601–17618 RSC.
- J. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- M. Moliner, C. Martínez and A. Corma, Chem. Mater., 2013, 26, 246–258 CrossRef.
- R. C. Weast, CRC Handbook of Chemistry and Physics, CRC Press, 1967 Search PubMed.
- B. Poling, J. Prausnitz and J. O. Connell, The Properties of Gases and Liquids, McGraw-Hill, 2000 Search PubMed.
- X. Xu, C. Song, R. Wincek, J. M. Andresen, B. G. Miller and A. W. Scaroni, Prepr. Symp. - Am. Chem. Soc., Div. Fuel Chem., 2003, 48, 162–163 CAS.
- R. M. Barrer, Discuss. Faraday Soc., 1949, 7, 135–141 RSC.
- R. M. Barrer, J. Colloid Interface Sci., 1966, 21, 415–434 CrossRef CAS.
- R. M. Barrer and D. W. Brook, Trans. Faraday Soc., 1953, 49, 1049–1059 RSC.
- R. M. Barrer and D. W. Brook, Trans. Faraday Soc., 1953, 49, 940–948 RSC.
- R. M. Barrer and B. E. F. Fender, J. Phys. Chem. Solids, 1961, 21, 1–11 CrossRef CAS.
- R. M. Barrer and L. V. Rees, Trans. Faraday Soc., 1954, 50, 852–863 RSC.
- R. M. Barrer and D. W. Riley, J. Chem. Soc., 1948, 133–143 RSC.
- R. M. Barrer and A. B. Robins, Trans. Faraday Soc., 1953, 49, 929–939 RSC.
- R. M. Barrer and D. W. Riley, Trans. Faraday Soc., 1950, 46, 853–861 RSC.
- R. M. Barrer and D. A. Ibbitson, Trans. Faraday Soc., 1944, 40, 195–206 RSC.
- R. M. Barrer, Proc. R. Soc. London, A, 1938, 167, 392–420 CrossRef.
- S. S. Shepelev and K. G. Ione, React. Kinet. Catal. Lett., 1983, 23, 319–322 CrossRef CAS.
- M. Minachev Kh and I. Isakov Ya, in Molecular Sieves, American Chemical Society, 1973, pp. 451–460 Search PubMed.
- I. I. Ivanova and E. E. Knyazeva, Chem. Soc. Rev., 2013, 42, 3671–3688 RSC.
- A. Hedström, J. Environ. Eng., 2001, 127, 673–681 CrossRef.
- S. E. Bailey, T. J. Olin, R. M. Bricka and D. D. Adrian, Water Res., 1999, 33, 2469–2479 CrossRef CAS.
- S. Wang and Y. Peng, Chem. Eng. J., 2010, 156, 11–24 CrossRef CAS.
- P. Misaelides, Microporous Mesoporous Mater., 2011, 144, 15–18 CrossRef CAS.
- N. Hedin, L. Andersson, L. Bergström and J. Yan, Appl. Energy, 2013, 104, 418–433 CrossRef CAS.
- F. N. Ridha and P. A. Webley, Sep. Purif. Technol., 2009, 67, 336–343 CrossRef CAS.
- Q. Liu, A. Mace, Z. Bacsik, J. Sun, A. Laaksonen and N. Hedin, Chem. Commun., 2010, 46, 4502–4504 RSC.
- T.-H. Bae, M. R. Hudson, J. A. Mason, W. L. Queen, J. J. Dutton, K. Sumida, K. J. Micklash, S. S. Kaye, C. M. Brown and J. R. Long, Energy Environ. Sci., 2013, 6, 128–138 CAS.
- Z. Liu, C. A. Grande, P. Li, J. Yu and A. E. Rodrigues, Sep. Sci. Technol., 2011, 46, 434–451 CrossRef CAS.
- M. M. Lozinska, E. Mangano, J. P. S. Mowat, A. M. Shepherd, R. F. Howe, S. P. Thompson, J. E. Parker, S. Brandani and P. A. Wright, J. Am. Chem. Soc., 2012, 134, 17628–17642 CrossRef CAS PubMed.
- M. Palomino, A. Corma, J. L. Jorda, F. Rey and S. Valencia, Chem. Commun., 2012, 48, 215–217 RSC.
- Y. Cui, H. Kita and K. Okamoto, J. Mater. Chem., 2004, 14, 924–932 RSC.
- Q. Jiang, J. Rentschler, G. Sethia, S. Weinman, R. Perrone and K. Liu, Chem. Eng. J., 2013, 230, 380–388 CrossRef CAS.
- Q. Liu, T. Pham, M. D. Porosoff and R. F. Lobo, ChemSusChem, 2012, 5, 2237–2242 CrossRef CAS PubMed.
- O. Cheung, Q. Liu, Z. Bacsik and N. Hedin, Microporous Mesoporous Mater., 2012, 156, 90–96 CrossRef CAS.
- M. Castro, R. Garcia, S. J. Warrender, A. M. Z. Slawin, P. A. Wright, P. A. Cox, A. Fecant, C. Mellot-Draznieks and N. Bats, Chem. Commun., 2007, 3470–3472 RSC.
- M. Hong, S. Li, H. F. Funke, J. L. Falconer and R. D. Noble, Microporous Mesoporous Mater., 2007, 106, 140–146 CrossRef CAS.
- M. E. Rivera-Ramos, G. J. Ruiz-Mercado and A. J. Hernández-Maldonado, Ind. Eng. Chem. Res., 2008, 47, 5602–5610 CrossRef CAS.
- Q. Liu, N. C. O. Cheung, A. E. Garcia-Bennett and N. Hedin, ChemSusChem, 2011, 4, 91–97 CrossRef CAS PubMed.
- R. M. Barrer, J. Chem. Soc., 1948, 127–132 RSC.
- D. W. Breck, Zeolite molecular sieves: structure, chemistry, and use, Wiley, 1973 Search PubMed.
- D. T. Hayhurst, Chem. Eng. Commun., 1980, 4, 729–735 CrossRef CAS.
- J. Janák, M. Krejčí and E. E. Dubský, Ann. N. Y. Acad. Sci., 1959, 72, 731–738 CrossRef.
- J. Zhang, R. Singh and P. A. Webley, Microporous Mesoporous Mater., 2008, 111, 478–487 CrossRef CAS.
- F. N. Ridha, Y. Yang and P. A. Webley, Microporous Mesoporous Mater., 2009, 117, 497–507 CrossRef CAS.
- T. Inui, Y. Okugawa and M. Yasuda, Ind. Eng. Chem. Res., 1988, 27, 1103–1109 CrossRef CAS.
- G. C. Watson, N. K. Jensen, T. E. Rufford, K. I. Chan and E. F. May, J. Chem. Eng. Data, 2011, 57, 93–101 CrossRef.
- J. Shang, G. Li, R. Singh, Q. Gu, K. M. Nairn, T. J. Bastow, N. Medhekar, C. M. Doherty, A. J. Hill, J. Z. Liu and P. A. Webley, J. Am. Chem. Soc., 2012, 134, 19246–19253 CrossRef CAS PubMed.
- J. Shang, G. Li, R. Singh, P. Xiao, J. Z. Liu and P. A. Webley, J. Phys. Chem. C, 2013, 117, 12841–12847 CAS.
- D. W. Breck, W. G. Eversole, R. M. Milton, T. B. Reed and T. L. Thomas, J. Am. Chem. Soc., 1956, 78, 5963–5972 CrossRef CAS.
- R. J. Harper, G. R. Stifel and R. B. Anderson, Can. J. Chem., 1969, 47, 4661–4670 CrossRef CAS.
- M. Palomino, A. Corma, F. Rey and S. Valencia, Langmuir, 2009, 26, 1910–1917 CrossRef PubMed.
- Y. Delaval and E. C. de Lara, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 869–877 RSC.
- O. Cheung, Z. Bacsik, Q. Liu, A. Mace and N. Hedin, Appl. Energy, 2013, 112, 1326–1336 CrossRef CAS.
- A. Mace, N. Hedin and A. Laaksonen, J. Phys. Chem. C, 2013, 117, 24259–24267 CAS.
- A. V. Larin, A. Mace, A. A. Rybakov and A. Laaksonen, Microporous Mesoporous Mater., 2012, 162, 98–104 CrossRef CAS.
- A. Mace, K. Laasonen and A. Laaksonen, Phys. Chem. Chem. Phys., 2014, 16, 166–172 RSC.
- E. Jaramillo and M. Chandross, J. Phys. Chem. B, 2004, 108, 20155–20159 CrossRef CAS.
- D. M. Ruthven, Microporous Mesoporous Mater., 2012, 162, 69–79 CrossRef CAS.
- D. M. Ruthven, Adsorption, 2001, 7, 301–304 CrossRef CAS.
- H. Yucel and D. M. Ruthven, J. Colloid Interface Sci., 1980, 74, 186–195 CrossRef CAS.
- H. Yucel and D. M. Ruthven, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 60–70 RSC.
- E. F. Kondis and J. S. Dranoff, Ind. Eng. Chem. Process Des. Dev., 1971, 10, 108–114 CAS.
- S. Araki, Y. Kiyohara, S. Tanaka and Y. Miyake, J. Colloid Interface Sci., 2012, 388, 185–190 CrossRef CAS PubMed.
- Y. Lee, J. A. Hriljac, T. Vogt, J. B. Parise, M. J. Edmondson, P. A. Anderson, D. R. Corbin and T. Nagai, J. Am. Chem. Soc., 2001, 123, 8418–8419 CrossRef CAS PubMed.
- D. R. Corbin, L. Abrams, G. A. Jones, M. M. Eddy, G. D. Stucky and D. E. Cox, J. Chem. Soc., Chem. Commun., 1989, 42–43 RSC.
- D. R. Corbin, L. Abrams, G. A. Jones, M. M. Eddy, W. T. A. Harrison, G. D. Stucky and D. E. Cox, J. Am. Chem. Soc., 1990, 112, 4821–4830 CrossRef CAS.
- T. M. Nenoff, J. B. Parise, G. A. Jones, L. G. Galya, D. R. Corbin and G. D. Stucky, J. Phys. Chem., 1996, 100, 14256–14264 CrossRef CAS.
- J. B. Parise and D. E. Cox, J. Phys. Chem., 1984, 88, 1635–1640 CrossRef CAS.
- J. B. Parise, L. Abrams, J. D. Jorgensen and E. Prince, J. Phys. Chem., 1984, 88, 2303–2307 CrossRef CAS.
- R. M. Barrer and E. V. T. Murphy, J. Chem. Soc. A, 1970, 2506–2514 RSC.
- G. Aguilar-Armenta, G. Hernandez-Ramirez, E. Flores-Loyola, A. Ugarte-Castaneda, R. Silva-Gonzalez, C. Tabares-Munoz, A. Jimenez-Lopez and E. Rodriguez-Castellon, J. Phys. Chem. B, 2001, 105, 1313–1319 CrossRef CAS.
- R. W. Triebe and F. H. Tezel, Gas Sep. Purif., 1995, 9, 223–230 CrossRef CAS.
- K. P. Lillerud and J. H. Raeder, Zeolites, 1986, 6, 474–483 CrossRef CAS.
- W. Xingqiao and X. Ruren, Stud. Surf. Sci. Catal., 1985, 24, 111–118 CrossRef.
- G. T. Kerr, Science, 1963, 140, 1412 CAS.
- T. Remy, S. A. Peter, L. Van Tendeloo, S. Van der Perre, Y. Lorgouilloux, C. E. A. Kirschhock, G. V. Baron and J. F. M. Denayer, Langmuir, 2013, 29, 4998–5012 CrossRef CAS PubMed.
- T. R. Cannan, E. M. Flanigen, R. T. Gajek, B. M. Lok, C. A. Messina and R. L. Patton, US Pat. US4440871A, Union Carbide Corporation, United States, 1982.
- B. M. Lok, C. A. Messina, R. L. Patton, R. T. Gajek, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1984, 106, 6092–6093 CrossRef CAS.
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146–1147 CrossRef CAS.
- R. Roldán, M. Sánchez-Sánchez, G. Sankar, F. J. Romero-Salguero and C. Jiménez-Sanchidrián, Microporous Mesoporous Mater., 2007, 99, 288–298 CrossRef.
- X. T. Ren, N. Li, J. Q. Cao, Z. Y. Wang, S. Y. Liu and S. H. Xiang, Appl. Catal., A, 2006, 298, 144–151 CrossRef CAS.
- E. Aubert, F. Porcher, M. Souhassou and C. Lecomte, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 687–700 Search PubMed.
- J. Jänchen and H. Stach, Energy Procedia, 2012, 30, 289–293 CrossRef.
- S. Wilson and P. Barger, Microporous Mesoporous Mater., 1999, 29, 117–126 CrossRef CAS.
- S. G. Hedge, P. Ratnasamy, L. M. Kustov and V. B. Kazansky, Zeolites, 1988, 8, 137–141 CrossRef CAS.
- V. R. Choudhary and S. Mayadevi, Langmuir, 1996, 12, 980–986 CrossRef CAS.
- Q. J. Chen, M. A. Springuel-Huet and J. Fraissard, Chem. Phys. Lett., 1989, 159, 117–121 CrossRef CAS.
- M. A. Carreon, S. Li, J. L. Falconer and R. D. Noble, J. Am. Chem. Soc., 2008, 130, 5412–5413 CrossRef CAS PubMed.
- S. R. Venna and M. A. Carreon, Langmuir, 2011, 27, 2888–2894 CrossRef CAS PubMed.
- A. G. Arévalo-Hidalgo, J. A. Santana, R. Fu, Y. Ishikawa and A. J. Hernández-Maldonado, Microporous Mesoporous Mater., 2010, 130, 142–153 CrossRef.
- M. Briend, R. Vomscheid, M. J. Peltre, P. P. Man and D. Barthomeuf, J. Phys. Chem., 1995, 99, 8270–8276 CrossRef CAS.
- T. Takeguchi, W. Tanakulrungsank and T. Inui, Gas Sep. Purif., 1993, 7, 3–9 CrossRef CAS.
- S. Li, J. L. Falconer and R. D. Noble, J. Membr. Sci., 2004, 241, 121–135 CrossRef CAS.
- J. C. Poshusta, V. A. Tuan, E. A. Pape, R. D. Noble and J. L. Falconer, AIChE J., 2000, 46, 779–789 CrossRef CAS.
- S. Li, G. Alvarado, R. D. Noble and J. L. Falconer, J. Membr. Sci., 2005, 251, 59–66 CrossRef CAS.
- S. Li, J. G. Martinek, J. L. Falconer, R. D. Noble and T. Q. Gardner, Ind. Eng. Chem. Res., 2005, 44, 3220–3228 CrossRef CAS.
- S. Li, J. L. Falconer, R. D. Noble and R. Krishna, Ind. Eng. Chem. Res., 2006, 46, 3904–3911 CrossRef.
- X. Su, P. Tian, D. Fan, Q. Xia, Y. Yang, S. Xu, L. Zhang, Y. Zhang, D. Wang and Z. Liu, ChemSusChem, 2013, 6, 911–918 CrossRef CAS PubMed.
- M. L. Carreon, S. Li and M. A. Carreon, Chem. Commun., 2012, 48, 2310–2312 RSC.
- I. Deroche, L. Gaberova, G. Maurin, P. Llewellyn, M. Castro and P. Wright, Adsorption, 2008, 14, 207–213 CrossRef CAS.
- H. Maghsoudi, M. Soltanieh, H. Bozorgzadeh and A. Mohamadalizadeh, Adsorption, 2013, 19, 1045–1053 CrossRef CAS.
- S. Himeno, T. Tomita, K. Suzuki, K. Nakayama, K. Yajima and S. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 6989–6997 CrossRef CAS.
- M. Miyamoto, Y. Fujioka and K. Yogo, J. Mater. Chem., 2012, 22, 20186–20189 RSC.
- J. van den Bergh, W. Zhu, J. C. Groen, F. Kapteijn, J. A. Moulijn, K. Yajima, K. Nakayama, T. Tomita and S. Yoshida, in Stud. Surf. Sci. Catal., ed. Z. G. J. C. Ruren Xu and Y. Wenfu, Elsevier, 2007, pp. 1021–1027 Search PubMed.
- J. van den Bergh, W. Zhu, J. Gascon, J. A. Moulijn and F. Kapteijn, J. Membr. Sci., 2008, 316, 35–45 CrossRef CAS.
- T. D. Pham, R. Xiong, S. I. Sandler and R. F. Lobo, Microporous Mesoporous Mater., 2014, 185, 157–166 CrossRef CAS.
- A. Philippou and M. W. Anderson, Zeolites, 1996, 16, 98–107 CrossRef CAS.
- C. B. Lopes, M. Otero, J. Coimbra, E. Pereira, J. Rocha, Z. Lin and A. Duarte, Microporous Mesoporous Mater., 2007, 103, 325–332 CrossRef CAS.
- C. B. Lopes, E. Pereira, Z. Lin, P. Pato, M. Otero, C. M. Silva, J. Rocha and A. C. Duarte, Microporous Mesoporous Mater., 2011, 145, 32–40 CrossRef CAS.
- P. J. E. Harlick and F. H. Tezel, Microporous Mesoporous Mater., 2004, 76, 71–79 CrossRef CAS.
- S. M. Kuznicki, V. A. Bell, S. Nair, H. W. Hillhouse, R. M. Jacubinas, C. M. Braunbarth, B. H. Toby and M. Tsapatsis, Nature, 2001, 412, 720–724 CrossRef CAS PubMed.
- V. A. Bell, D. R. Anderson, B. K. Speronello, M. Rai and W. B. Dolan, US Pat. US2009004084A1, 2006.
- S. M. Kuznicki, US Pat. US07449023, Engelhard Corporation, United States, 1989.
- S. W. Park, S. H. Cho, W. S. Ahn and W. J. Kim, Microporous Mesoporous Mater., 2011, 145, 200–204 CrossRef CAS.
- A. Anson, C. C. H. Lin, S. M. Kuznicki and J. A. Sawada, Chem. Eng. Sci., 2009, 64, 3683–3687 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |