
One-step chemoselective conversion of tetrahydropyranyl ethers to silyl-protected alcohols†
Abstract
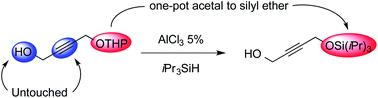
Aluminium trichloride catalyses the expeditious direct conversion of tetrahydropyranyl ethers to silyl ethers. This one-step transformation is chemoselective versus deprotection of the acetal and hydrosilylation of unsaturated carbon–carbon bonds, and can also be applied to linear acetals. A possible mechanism is tentatively proposed.
 Please wait while we load your content...
Please wait while we load your content...

