Intramolecular cyclization–decyclization of new sterically hindered diiminophenol. Synthesis and coordination abilities†
Gleb A. Abakumovab,
Nikolay O. Druzhkova,
Elena N. Egorova*a,
Tatiana N. Kocherovaa,
Andrey S. Shavyrina and
Anton V. Cherkasova
aG.A. Razuvaev Institute of Organometallic Chemistry RAS, Tropinina str. 49, 603950, Nizhny Novgorod, Russia. E-mail: ee@iomc.ras.ru; Fax: +7 8312 4627497; Tel: +7 8312 4627682
bLobachevsky State University of Nizhny Novgorod, Gagarin Ave. 23/5, 603950, Nizhny Novgorod, Russia
First published on 7th March 2014
Abstract
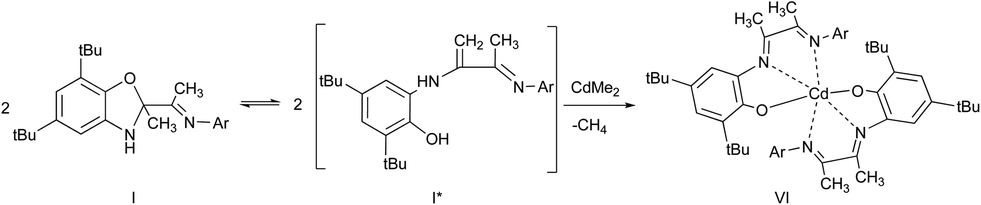
The new sterically hindered benzoxazole I was synthesized by the reaction of 3-(2,6-diisopropylphenylimino)butan-2-one and 2-amino-4,6-di-tert-butylphenol. It is shown that compound I is in equilibrium with the open enamine form in solution. The coordination abilities of I have been studied. The ligand I is shown to demonstrate either a neutral coordination type in complex with cadmium iodide or monoanionic type in cadmium complexes obtained by the interaction of I with Me2Cd.
Introduction
Schiff base ligands have been extensively studied mainly due to their coordination abilities, facile syntheses, easily tunable steric factors, electronic properties and good solubility in common solvents.1–4 Transition metal complexes with oxygen and nitrogen donor Schiff bases (O,N-ligands) are of particular interest1–4 because of their ability to possess unusual configurations and to be structurally labile. The Fe(III), Co(III), Ni(II), Zn(II), Cd(II) and lanthanide(III) complexes with O,N,N-ligand synthesized by reaction of 2-pyridincarboxaldehyde and substituted aminophenols5 have been obtained and investigated.6 Some of them can be used as photoactive materials.6–12 In a series of papers it have been shown that the iminophenol derivatives synthesized by condensation of o-aminophenols with various substituted aldehydes can undergo the intramolecular cyclization to give five- or six-membered rings13–16 opening in alkaline solution in presence of metal ion.17Recently we have described the synthesis of iminoketone,18 which can act as carbonyl reagent in condensation reactions with substituted o-aminophenols. In this case the resulting products feature hydroxyl group and sterically hindered N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CN fragment. Such compounds may be used as either neutral or valent bonded ligands.
C–CN fragment. Such compounds may be used as either neutral or valent bonded ligands.
Results and discussion
This study is aimed to the synthesis of the new sterically hindered O,N,N-ligand derived from the interaction of 3-(2,6-diisopropylphenylimino)butan-2-one with 2-amino-4,6-di-tert-butylphenol and the investigation of the properties of obtained compound. The treatment of 3-(2,6-diisopropylphenylimino)-butan-2-one with the o-aminophenol leads to the colorless crystalline solid compound identified as dihydrobenzoxazole I by the spectral methods, 15N–1H 2D ge-HSQC NMR spectrum and the X-ray structural analysis (Fig. 1). Compound I is the product of intramolecular cyclization of the desired N-substituted o-iminophenol (Scheme 1).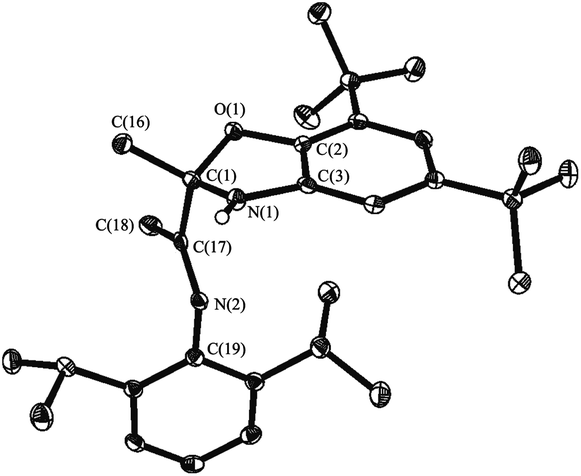 | ||
| Fig. 1 The molecular structure of I. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms (except bonded to N(1)) are omitted for clarity. Selected distances [Å] and angles [°]: C(1)–O(1) 1.464(3), C(1)–N(1) 1.464(3), C(2)–O(1) 1.393(3), C(3)–N(1) 1.409(3), C(1)–C(16) 1.514(3), C(1)–C(17) 1.534(3), C(17)–C(18) 1.500(3), C(17)–N(2) 1.276(3), N(2)–C(19) 1.423(3), C(1)–O(1)–C(2) 105.7(2), C(1)–N(1)–C(3) 105.2(2), C(17)–N(2)–C(19) 122.0(2). | ||
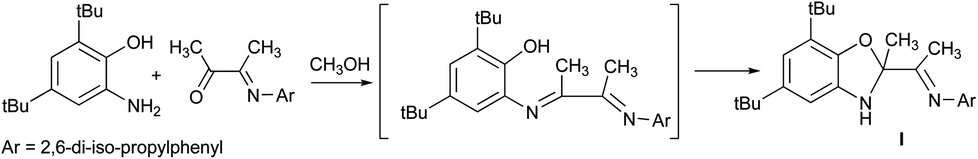 | ||
| Scheme 1 The synthesis of compound I. | ||
As we have mentioned above the o-iminophenols based on o-aminophenols with various substituted aldehydes are in equilibrium with an isomeric cyclic form of ligands. We tried to indicate each form of I by 1H NMR in various solvents, but unfortunately concentration of this form is too low to be detected by NMR. This fact displays that the equilibrium is shifted completely to the dihydrobenzoxazole. However, an interesting phenomenon took place in deuterium methanol solution of I.
The freshly prepared solution of compound I in CD3OD features 1H NMR typical for such compounds. However, within 3–5 minutes in solution the intensity of the Csp3–CH3 methyl group signal is greatly reduced (Fig. 2). In 13C NMR spectrum signal of this methyl group is transformed into a multiplet (Fig. 3).
 | ||
| Fig. 2 Fragment of 1H NMR spectrum of I in CD3OD: (a) freshly prepared solution; and (b) after 5 min. | ||
 | ||
| Fig. 3 Fragment of 13C NMR spectrum of I in CD3OD after 5 min. | ||
The decreasing of the methyl group signal intensity and arising of multiplet in 13C NMR can be caused by methyl group deuteration. The fact of selective deuteration of OH and NH groups in methanol-d4 and some other NMR solvents is widely known but selective deuteration of the methyl group in mild conditions is absolutely unusual.
Supposed H–D exchange can be explained by cyclization–decyclization equilibrium in solution (Scheme 2). The hydroxyl of I* is rapidly deuterated in CD3OD forming OD group. Subsequently, deuterium of OD group migrates into methyl group forming a partially deuterated CH2D group and then CHD2 group up to fully deuterated CD3 group (compound II).
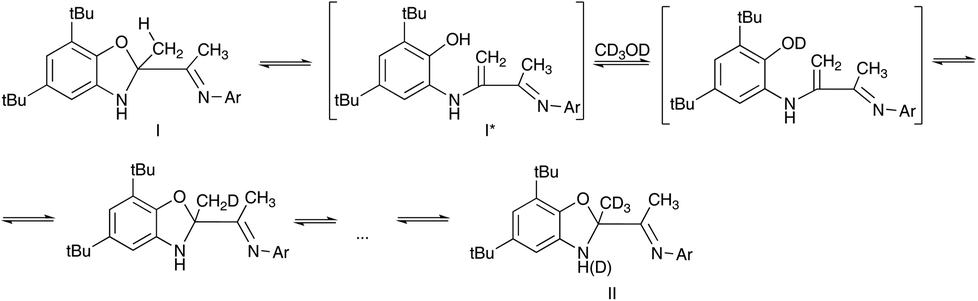 | ||
| Scheme 2 The deuteration process in CD3OD solution. | ||
It should be noted that the peak of NH group is still present in the 1H NMR spectrum while the Csp3–CH3 methyl group signal is completely disappear.
After a three-time recrystallization of I in CD3OD the selectively deuterated product II was isolated. The deuteration degree is proven by absence of the methyl group signal in 1H NMR spectrum. There are three signals observed in 2H NMR spectrum of compound II. The chemical shift of the most intensive signal assigned to CD3 group (1.80 ppm) is close to the chemical shift of the methyl protons in source compound I (1.83 ppm). The intensity of 2H signals of CHD2 (1.81 ppm) and CH2D (1.82 ppm) groups are substantially lower.
The mass spectrum of II showed peaks both for the molecular ion at m/z = 451 and the ion corresponding to fragment of the molecule containing benzoxazole rings (m/z = 249) while the source compound I shows peaks at m/z = 449 (M+) and 246.
Taking into account the reaction conditions, the multiplicity of methyl signal in 13C NMR and the intensity of residual methyl protons in 1H NMR spectra we may affirm that the deuteration mostly leads to product with completely deuterated methyl group.
Another evidence of the existence of enamine form I* in solution is the oxidation of I by alkaline solution of potassium ferricyanide (Scheme 3). In this case Csp3–Me group undergoes oxidation and the benzoxazine derivative III is formed. This reaction is possible due to the ring opening of I with formation of I* intermediate.
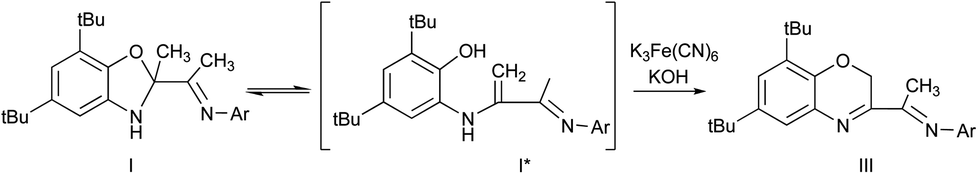 | ||
| Scheme 3 The synthesis of compound III. | ||
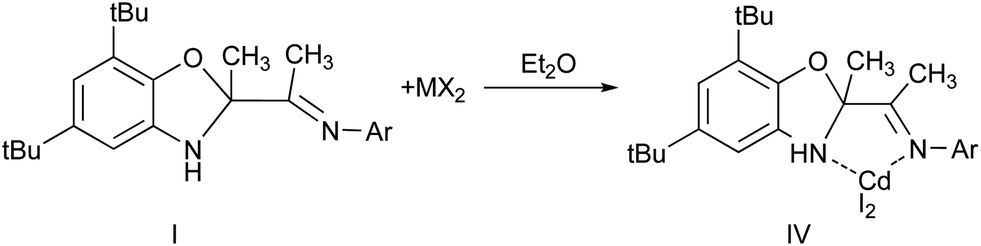
The coordination abilities of the ligand I have been investigated. It is known that the neutral complexes of R–DAB (N,N′-disubstituted diazabutadienes) are prepared by mixing of metal salt with the R–DAB ligand in stoichiometric molar ratio.19 In our case the interaction of ligand I with cadmium iodide results in formation of colorless powder of metal complex IV (Scheme 4).
 | ||
| Scheme 4 The synthesis of compound IV. | ||
The signals shifting in NMR spectrum of obtained complex IV in comparison with source ligand I indicates the electron density displacement from organic ligand to metal atom and formation of molecular complex L*CdI2 (L = I). In this case I acts as a neutral ligand coordinated by two nitrogen atoms.
The structure of IV has been determined by single-crystal X-ray diffraction (Fig. 4). Cadmium atom in IV is in distorted tetragonal coordination environment with two nitrogen and two iodide atoms on the tops. Dihedral angle between two aromatic rings is slightly more than observed for the parent ligand I (86.0°) amounts to 88.7°. The distances Cd(1)–N(1) (2.347(1) Å) and Cd(1)–N(2) (2.297(2) Å) are shorter than the sum of van der Waals radii of cadmium and nitrogen atoms (3.7 Å), and are slightly more than the sum of covalent radii of these atoms (2.1 Å (ref. 20)). So the Cd–N distances are in the typical range of donor–acceptor bond lengths between aforementioned atoms. The bond lengths C(15)–O(1) (1.456(2) Å), C(15)–N(1) (1.469(2) Å) and C(17)N(2) (1.275(2) Å) in ligand are in the range expected for organic compounds.
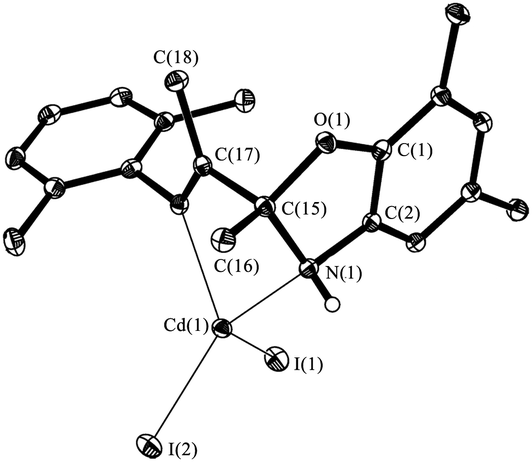 | ||
| Fig. 4 The molecular structure of IV. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms (except bonded to N(1)) and Me-fragments of iPr-, tBu-groups are omitted. Selected distances [Å] and angles [°]: Cd(1)–N(1) 2.347(1), Cd(1)–N(2) 2.297(2), Cd(1)–I(1) 2.6714(2), Cd(1)–I(2) 2.7132(2), O(1)–C(1) 1.393(2), O(1)–C(15) 1.456(2), N(1)–C(2) 1.451(2), N(1)–C(15) 1.469(2), C(1)–C(2) 1.378(2), C(15)–C(17) 1.543(2), C(15)–C(16) 1.512(2), C(17)–C(18) 1.494(3), N(2)–C(17) 1.275(2), N(2)–C(19) 1.453(2), I(1)–Cd(1)–I(2) 120.395(2), N(1)–Cd(1)–N(2) 71.48(5), C(1)–O(1)–C(15) 104.5(1), C(2)–N(1)–C(15) 102.3(1), C(17)–N(2)–C(19) 120.1(2). | ||
In spite of I has cyclic structure in solid state methyl group deuteration in CD3OD solution means that the open form (I*) containing phenol group is present in solution.
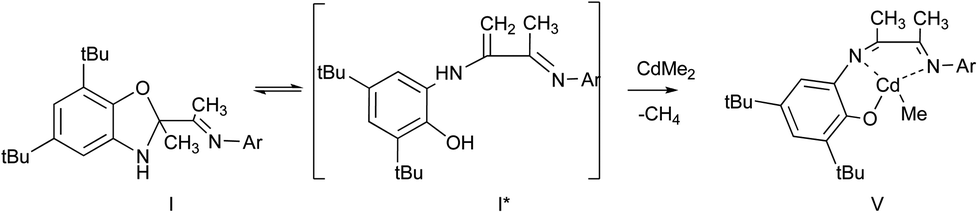
The interaction of I with equimolar amount of dimethylcadmium in ether solution leads to cadmium phenolate derivative V. The reaction is accompanied with solution color change and release of methane. After cooling of the reaction mixture the deep brown crystals V were isolated with yield 82% (Scheme 5).
 | ||
| Scheme 5 The synthesis of compound V. | ||
The molecular structure of V is depicted in Fig. 5. According to X-ray data analysis V adopts a dimeric structure with two cadmium cations bounded by two bridging oxygen atoms. The Cd(1) is in distorted tetragonal pyramidal environment: O(1), N(1), N(2) and C(61) form the base while O(2) occupies an apical site. The Cd(2) is in distorted tetrahedron environment with the O(1), O(2), N(3) and C(62) in the tops. The Cd(1), O(1), Cd(2) and O(2) form a distorted rhombus. The Cd(1)–O(1) (2.374(3) Å) and Cd(2)–O(2) (2.225(3) Å)21,22 distances are significantly shorter than bonds Cd(1)–O(2) and Cd(2)–O(1) (2.398(3) Å and 2.240(2) Å) which have donor–acceptor nature. Also these distances are shorter than the sum of covalent radii of these atoms. Values of Cd(1)–N(1) (2.344(3) Å), Cd(1)–N(2) (2.428(4) Å) and Cd(2)–N(3)(2.578(3) Å) lie in the range typical for donor–acceptor bond lengths of aforementioned atoms. The cadmium atoms separated from each other by 3.370(4) Å. The distance between Cd(2) and N(4) atoms is 5.305(3) Å. This fact demonstrates that the N(4) atom is not coordinated on metal atom.
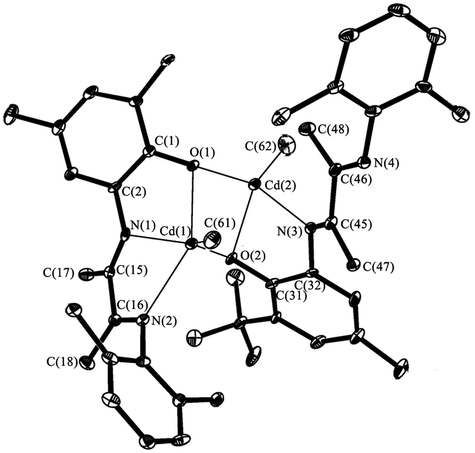 | ||
| Fig. 5 The molecular structure of V. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms and Me-fragments of iPr-, tBu-groups are omitted. Selected distances [Å] and angles [°]: Cd(1)–O(1) 2.374(3), Cd(1)–N(1) 2.344(3), Cd(1)–N(2) 2.428(4), Cd(1)–C(61) 2.146(4), Cd(2)–O(2) 2.225(3), Cd(2)–N(3) 2.578(3), Cd(2)–C(62) 2.146(5), O(1)–C(1) 1.325(5), C(1)–C(2) 1.434(6), N(1)–C(2) 1.402(5), N(1)–C(15) 1.289(5), N(2)–C(16) 1.274(5), N(2)–C(19) 1.441(5), C(1)–C(2) 1.434(6), C(15)–C(16) 1.520(5), C(15)–C(17) 1.508(6), C(16)–C(18) 1.507(6), O(2)–C(31) 1.361(5), N(3)–C(32) 1.434(6), N(3)–C(45) 1.285(5), N(4)–C(46) 1.271(5), N(4)–C(49) 1.426(5), C(31)–C(32) 1.406(6), C(45)–C(46) 1.500(6), C(45)–C(47) 1.517(5), C(46)–C(48) 1.514(5), O(1)–Cd(1)–N(1) 62.7(1), N(1)–Cd(1)–N(2) 68.2(1), O(2)–Cd(2)–N(3) 67.9(1), O(1)–Cd(1)–O(2) 78.6(1), O(1)–Cd(2)–O(2) 85.2(2). | ||
In accordance to X-ray analysis ligands in dimer V are not identically coordinated. However there is only one set of signals belonging to the ligand in 1H NMR spectrum. The above data may be caused by either dissociation of V in solution or the coordination sphere dynamics.
The interaction of Me2Cd with I (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) leads to the formation of deep blue derivative VI (Scheme 6).
:2) leads to the formation of deep blue derivative VI (Scheme 6).
 | ||
| Scheme 6 The synthesis of compound VI. | ||
The 1H and 13C NMR spectra of VI demonstrate one set of signals belonging to the ligand.
The X-ray analysis of VI shows that the phenolate ligands with diazabutadiene fragments are identically coordinated (Fig. 6). The Cd(1) is in distorted octahedral environment. The N(1), N(3), N(4) and O(2) form the base while O(1) and N(2) occupy an apical sites. The o-aminophenolate fragments are plane and the dihedral angle between ones amounts 84.9°. The rings of aniline fragments of the ligand are almost parallel to each other. Dihedral angle between ones is 18.4°. The Cd(1)–O(1) (2.258(1) Å) and Cd(1)–O(2) (2.262(1) Å) distances are significantly shorter than bonds Cd–O which have donor–acceptor nature. These distances are comparable with Cd–O bonds lengths observed for the cadmium phenolate compounds.21,22 Values of Cd–N distances (Cd(1)–N(1) 2.333(1), Cd(1)–N(2) 2.397(1), Cd(1)–N(3) 2.318(1) and Cd(1)–N(4) 2.317(1) Å) lie in the range typical for donor–acceptor bond nature between aforementioned atoms. The distances C–C and C–N (C(15)–C(16) 1.510(2) Å, C(45)–C(46) 1.510(2) Å, N(1)–C(15) 1.286(2) Å, N(2)–C(17) 1.286(2) Å, N(3)–C(45) 1.289(2) Å and N(4)–C(46) 1.295 Å) are corresponded to bond orders of one and two, respectively.
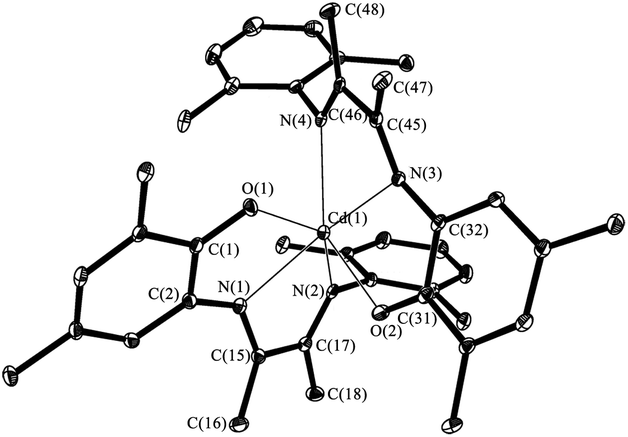 | ||
| Fig. 6 The molecular structure of VI. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms and Me-fragments of iPr-, tBu-groups are omitted. Selected distances [Å] and angles [°]: Cd(1)–O(1) 2.258(1), Cd(1)–O(2) 2.262(1), Cd(1)–N(1) 2.333(1), Cd(1)–N(2) 2.397(1), Cd(1)–N(3) 2.318(1), Cd(1)–N(4) 2.371(1), O(1)–C(1) 1.299(3), N(1)–C(2) 1.404(2), N(1)–C(15) 1.286(2), N(2)–C(17) 1.286(2), N(2)–C(19) 1.450(1), C(1)–C(2) 1.438(2), C(15)–C(16) 1.510(2), C(15)–C(17) 1.500(2), C(16)–C(18) 1.502(2), O(2)–C(31) 1.301(2), N(3)–C(32) 1.404(2), N(3)–C(45) 1.289(2), N(4)–C(46) 1.295(2), N(4)–C(49) 1.444(2), C(31)–C(32) 1.430(2), C(45)–C(46) 1.510(2), C(45)–C(47) 1.498(2), C(46)–C(48) 1.495(2), O(1)–Cd(1)–N(1) 72.14(4), N(1)–Cd(1)–N(2) 70.34(5), O(2)–Cd(1)–N(3) 72.48(4), N(3)–Cd(1)–N(4) 70.41(5), O(1)–Cd(1)–O(2) 96.46(4). | ||
Conclusions
In the present work we have described the synthesis of novel ligand–benzoxazole I. It is shown that obtained compound I undergoes a ring open process to form the enamine species in solution. The coordination abilities of I have been studied. The ligand I is shown to demonstrate a neutral coordination type in complex with cadmium halide. A convenient procedure for the synthesis of cadmium complexes with the new sterically hindered O,N,N-ligand has been developed. This method may be used for a synthesis of the similar complexes containing different metals.Experimental
General
2-Amino-4,6-di-tert-butylphenol was prepared according to a previously described procedures.23 Solvents were purified by standard methods.24 All synthesis have been conducted in evacuated ampoules.The NMR spectra were recorded on a “Bruker Avance III” NMR spectrometer (400 MHz) using CDCl3, CD3OD or C6D6 as the solvents and tetramethylsilane as the internal standard. IR-spectra were recorded by ‘Specord M-80. Elemental analyses were obtained on “EuroEA-3028-HT”. Mass spectra was recorded on mass spectrometer “Polaris Q”’ with ion trap mass analiser. Electron impact mass spectra (70 eV) were registrated in the mass range 50–550 m/z.
X-Ray crystallographic study of I, IV–VI
The X-ray data were collected on a Smart Apex diffractometer (for I and IV, graphite-monochromated, MoKα-radiation, ω-scan technique, λ = 0.71073 Å, T = 100(2) K) and a Agilent Xcalibur E diffractometer (for V and VI, graphite-monochromated, MoKα-radiation, ω-scan technique, λ = 0.71073 Å, T = 100(2) K). The structures were solved by direct methods and were refined on F2 using SHELXTL25 (I and IV) and CrysAlis Pro26 (V and VI) package. All non-hydrogen atoms were found from Fourier syntheses of electron density and were refined anisotropically. H1A in I was also found from Fourier syntheses of electron density, but were refined isotropically. All other hydrogen atoms were placed in calculated positions and were refined in the riding model. SADABS27 (I and IV) and ABSPACK (CrysAlis Pro)26 (V and VI) were used to perform area-detector scaling and absorption corrections. The details of crystallographic, collection and refinement data are shown in Table 1.†| I | IV | V | VI | |
|---|---|---|---|---|
| Formula | C30H44N2O | C34H54CdI2N2O2 | C68.20H107.50Cd2N4O3.55 | C64H96CdN4O3 |
| Mr | 448.67 | 888.99 | 1265.08 | 1081.85 |
| Crystal size, mm3 | 0.15 × 0.10 × 0.05 | 0.42 × 0.14 × 0.12 | 0.40 × 0.10 × 0.10 | 0.40 × 0.20 × 0.20 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2(1)/n | P |
P2(1)/n |
| a, Å | 11.2464(9) | 19.3052(6) | 12.7219(2) | 15.0475(3) |
| b, Å | 15.861(1) | 11.9215(4) | 17.4042(4) | 26.8007(4) |
| c, Å | 18.527(1) | 19.4653(7) | 17.4215(4) | 16.0102(3) |
| α, ° | 102.754(2) | 90 | 72.473(2) | 90 |
| β, ° | 107.317(2) | 119.466(1) | 78.284(2) | 109.913(2) |
| γ, ° | 109.440(2) | 90 | 73.561(2) | 90 |
| Cell volume, Å | 2781.0(4) | 3900.4(2) | 3498.5(1) | 6070.6(2) |
| Z | 4 | 4 | 2 | 4 |
| Dcalc, g cm−3 | 1.072 | 1.514 | 1.201 | 1.184 |
| μ, mm−1 | 0.064 | 2.171 | 0.652 | 0.405 |
| F000 | 984 | 1768 | 1338 | 2320 |
| 2θ range, ° | 52 | 52 | 52 | 52 |
| Index ranges | −13 ≤ h ≤ 13 | −23 ≤ h ≤ 23 | −15 ≤ h ≤ 15 | −18 ≤ h ≤ 18 |
| −19 ≤ k ≤ 19 | −14 ≤ k ≤ 14 | −21 ≤ k ≤ 21 | −33 ≤ k ≤ 33 | |
| −22 ≤ l ≤ 22 | −23 ≤ l ≤ 24 | −21 ≤ l ≤ 21 | −19 ≤ l ≤ 19 | |
| Reflns collected | 23923 |
32685 |
53781 |
92898 |
| Independent reflns | 10872 |
7604 | 13581 |
11878 |
| Rint | 0.0452 | 0.0211 | 0.0674 | 0.0907 |
| Completeness to θ | 99.6 | 99.4 | 98.7 | 99.6 |
| Data/restraints/parameters | 10872/0/619 |
7604/0/388 | 13581/45/736 |
11878/7/688 |
| GooF | 1.038 | 1.026 | 1.053 | 1.033 |
| R1 (I > 2σ(I)) | 0.0667 | 0.0229 | 0.0748 | 0.0392 |
| wR2 (all data) | 0.1528 | 0.0566 | 0.1943 | 0.1020 |
Synthesis
Yield: 0.57 g (85%); m.p. = 82 °C. m/z 448 (M+, 100%); 449 (M+ + 1, 33); 450 (M+ + 2, 6). Found (%): C, 80.4; H, 9.9. Calculated for C30H44N2O (%): C, 80.3; H, 9.9. IR (nujol, ν/cm−1): 3291br, 3062m, 1914w, 1857w, 1796w, 1666s, 1624w, 1605m, 1418s, 1365s, 1327m, 1300m, 1266m, 1224s, 1193s, 1105s, 1079s, 1029w, 1014w, 938w, 911w, 892m, 854s, 823m, 796w, 774s, 747m, 716m, 667m, 644w, 583w, 552w, 510w. 1H NMR (CDCl3, δ/ppm, J/Hz): 0.79 and 0.83 (both d, 6H, (CH3)2CH, J = 6.88); 1.26 and 1.36 (both s, 9H, tBu); 1.83 and 1.86 (both s, 3H, Me); 2.23 and 2.64 (both sept, 1H, (CH3)2CH, J = 6.88 Γ ); 5.88 (s, 1H, NH); 6.74 (br s, 2H, Harom); 7.02–7.14 (m, 3H, Harom). 1H NMR (CD3OD, δ/ppm, J/Hz): 0.81 and 0.85 (both d, 3H, (CH3)2CH, J = 6.88); 1.13 and 1.16 (both d, 3H, (CH3)2CH, J = 6.88); 1.27 and 1.35 (both s, 9H, tBu); 1.81 and 1.83 (both s, 3H, Me); 2.31 and 2.69 (both sept, 1H, (CH3)2CH, J = 6.88 Γ); 4.57 (s, 1H, NH); 6.73 and 6.75 (both d, 1H, Harom, J = 7.26 Γ); 6.98–7.17 (m, 3H, Harom). 13C NMR (CDCl3, δ/ppm): 15.4, 22.3, 22.7, 22.8, 23.0, 23.2, 24.8, 27.9, 28.2, 29.7, 31.8, 34.0, 34.6, 76.7, 77.0, 77.3, 100.5, 107.4, 115.0, 122.9, 123.0, 123.8, 130.8, 135.7, 136.0, 138.0, 144.2, 144.7, 144.9, 171.7 (CN). 13C NMR (CD3OD, δ/ppm): 14.4; 21.5; 21.8; 22.0; 22.2; 27.6; 27.9; 29.0; 30.9; 33.6 (CH3); 34.1 (CH3); 100.3; 107.1; 114.5; 122.6; 123.7; 130.5; 135.5; 135.8; 138.1; 144.1; 144.7; 144.9; 172.0 (CN). The crystals of I suitable for X-ray were obtained from CH3CN.
); 5.88 (s, 1H, NH); 6.74 (br s, 2H, Harom); 7.02–7.14 (m, 3H, Harom). 1H NMR (CD3OD, δ/ppm, J/Hz): 0.81 and 0.85 (both d, 3H, (CH3)2CH, J = 6.88); 1.13 and 1.16 (both d, 3H, (CH3)2CH, J = 6.88); 1.27 and 1.35 (both s, 9H, tBu); 1.81 and 1.83 (both s, 3H, Me); 2.31 and 2.69 (both sept, 1H, (CH3)2CH, J = 6.88 Γ); 4.57 (s, 1H, NH); 6.73 and 6.75 (both d, 1H, Harom, J = 7.26 Γ); 6.98–7.17 (m, 3H, Harom). 13C NMR (CDCl3, δ/ppm): 15.4, 22.3, 22.7, 22.8, 23.0, 23.2, 24.8, 27.9, 28.2, 29.7, 31.8, 34.0, 34.6, 76.7, 77.0, 77.3, 100.5, 107.4, 115.0, 122.9, 123.0, 123.8, 130.8, 135.7, 136.0, 138.0, 144.2, 144.7, 144.9, 171.7 (CN). 13C NMR (CD3OD, δ/ppm): 14.4; 21.5; 21.8; 22.0; 22.2; 27.6; 27.9; 29.0; 30.9; 33.6 (CH3); 34.1 (CH3); 100.3; 107.1; 114.5; 122.6; 123.7; 130.5; 135.5; 135.8; 138.1; 144.1; 144.7; 144.9; 172.0 (CN). The crystals of I suitable for X-ray were obtained from CH3CN.
Yield: 0.43 g (86%). m.p. 167 °C. Found (%): C, 80.7; H, 9.5. Calculated for C30H42N2O(%): C, 80.9; H, 9.3. IR (nujol, ν/cm−1): 1627m, 1589w, 1360s, 1315m, 1258m, 1243m, 1217m, 1089w, 1021m, 976w, 938w, 912w, 882m, 826w, 792w, 762s, 720w, 698w, 679w, 653w. 1H NMR (CDCl3, δ/ppm, J/Hz): 1.14 and 1.15 (both d, 6H, CH(CH3)2, J = 6.88); 1.34 (s, 9H, tBu); 1.41 (s, 9H, tBu); 2.07 (s, 3H, CH3); 2.63 (sept, 2H, CH(CH3)2, J = 6.88); 5.12 (s, 2H, CH2); 7.08–7.34 (m, 5H, Harom). 13C NMR (CDCl3, δ/ppm): 15.7; 22.7; 23.2; 28.3; 29.7; 31.5; 34.4; 34.7; 61.2 (CH2); 123.0; 123.2; 124.0; 124.6; 133.9; 135.2; 137.2; 143.6; 144.2; 145.8; 159.1 and 166.15 (CN).
Yield: 0.12 g (73%). Found (%): C, 44.2; H, 5.5; Cd, 13.7; I, 31.2. Calculated for C34H54CdI2N2O2 (%): C, 44.2; H, 5.4; Cd, 13.8; I, 31.2. IR (nujol, ν/cm−1): 3120s (N–H), 1646s (CN), 1413s, 1364s, 1326w, 1270m, 1236m, 1179s, 1134s, 1115s, 1081s, 965s, 916m, 893s, 871s, 837m, 803m, 788s, 754m, 743m, 720w, 626w. 1H NMR (CDCl3, δ/ppm, J/Hz): 0.45 (d, 3H, (CH3)2CH, J = 6.73); 0.96 (d, 3H, (CH3)2CH, J = 6.73); 1.19 (d, 3H, (CH3)2CH, J = 6.73); 1.25 (d, 3H, (CH3)2CH, J = 6.73); 1.33 (s, 9H, tBu); 1.35 (s, 9H, tBu); 1.96 (sept, 1H, (CH3)2CH, J = 6.73); 2.04 (s, 3H, CH3); 2.37 (s, 3H, CH3); 2.86 (sept, 1H, (CH3)2CH, J = 6.73); 4.85 (br s, 1H, NH); 7.04–7.25 (m, 5H, Harom). 13C NMR (CDCl3, δ/ppm): 17.98; 23.23; 23.77; 23.80; 24.15; 26.30; 28.22; 28.30; 29.91; 31.72; 34.42; 35.15; 100.54; 115.50; 121.96; 124.26; 124.43; 127.24; 131.15; 132.67; 138.08; 138.57; 140.12; 146.04; 146.28; 180.36 (CN). The crystals of IV suitable for X-ray were obtained from Et2O.
Yield: 1.024 g (89%). Found (%): C, 64.8; H, 8.0; Cd, 19.6. Calculated for C31H46CdN2O (%): C, 64.7; H, 8.1; Cd, 19.6. 1H NMR (CDCl3, δ/ppm, J/Hz): −0.69 (m, 3H, CH3Cd, JH–Cd = 82.47); 1.12 (d, 6H, (CH3)2CH, J = 6.86); 1.15 (d, 6H, (CH3)2CH, J = 6.86); 1.35 (s, 9H, tBu); 1.50 (s, 9H, tBu); 2.17 (s, 3H, CH3); 2.60 (sept, 2H, (CH3)2CH, J = 6.86); 2.64 (s, 3H, CH3); 6.87 (d, 1H, Harom, J = 2.48); 7.17–7.21 (m, 3H, Harom); 7.32 (d, 1H, Harom, J = 2.48). 13C NMR (CDCl3, δ/ppm): −13.65 (CH3Cd); 19.32; 23.75; 28.47; 29.55; 31.66; 34.00; 35.39; 115.65; 123.75; 124.83; 129.90; 131.44; 133.53; 137.47; 139.83; 142.50; 159.06 (CN); 162.73 (Carom–N); 167.48 (CN). The crystals of V suitable for X-ray were obtained from Et2O.
Yield: 0.84 g (83%). Found (%): C, 71.6; H, 8.6; Cd, 11.2. Calculated for C60H86CdN4O2 (%): C, 71.5; H, 8.6; Cd, 11.2. IR (nujol, ν/cm−1): 1618w, 1589w, 1556s, 1522m, 1506s, 1411m, 1373s, 1361s, 1325s, 1299m, 1278s, 1254s (C–O), 1189s, 1157s, 1121s, 1056w, 1024w, 979s, 935w, 908m, 870m, 837s, 793m, 781s, 734m, 704w, 645w, 633w, 597w, 583s, 553w, 512w, 485m. 1H NMR (C6D6, δ/ppm, J/Hz): 0.30 (d, 3H, (CH3)2CH, J = 6.81); 0.85 (d, 3H, (CH3)2CH, J = 6.81); 1.08–1.10 (m, 6H, (CH3)2CH, J = 6.81); 1.36 (s, 9H, tBu); 1.54 (s, 9H, tBu); 1.64 (s, 3H, CH3); 2.36 (sept, 1H, (CH3)2CH, J = 6.81); 2.41 (s, 3H, CH3); 3.58 (sept, 1H, (CH3)2CH, J = 6.81); 6.96 (d, 1H, Harom, J = 2.22); 6.87–7.03 (m, 3H, Harom); 7.44 (d, 1H, Harom, J = 2.22). 13C NMR (C6D6, δ/ppm): 19.50; 19.82; 22.75; 23.01; 24.11; 24.32; 27.39; 28.02; 29.58; 31.61; 33.82; 35.51; 116.69; 123.43; 124.53; 125.36; 125.86; 129.88; 130.54; 137.79; 139.13; 139.87; 145.13; 150.16; 165.68 (CN); 171.68 (CN). The crystals of VI suitable for X-ray were obtained from Et2O.
Acknowledgements
This work was supported by the Council for Grants of the President of Russian Federation for Support of Leading Scientific Schools (grant no. NSh 271.2014.3), the Russian Foundation for Basic Research (project no. 14-03-31071_mol_a).Notes and references
- G. van Koten and K. Vrieze, Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Acad. Press, New York, London, 1982, vol. 21, p. 152 Search PubMed.
- L. F. Lindoy and S. E. Livingstone, Coord. Chem. Rev., 1967, 2, 173 CrossRef CAS.
- P. Preishuber-Pflugl and M. Brookhart, Macromolecules, 2002, 35, 6074 CrossRef CAS.
- D. J. Tempel, L. K. Johnson, R. Leigh Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686 CrossRef CAS.
- A. Majumder, G. Rosair, A. Mallick, N. Chattopadhyay and S. Mitra, Polyhedron, 2006, 25, 1753 CrossRef CAS PubMed.
- G. Juhasz, S. Hayami, O. Sato and Y. Maeda, Chem. Phys. Lett., 2002, 364, 164 CrossRef CAS.
- S. Hayami and Y. Maeda, Inorg. Chim. Acta, 1997, 255, 181 CrossRef CAS.
- J. R. Dilworth, S. D. Howe, D. J. Hutson, J. R. Miller, J. Silver, R. M. Thompson, M. Harman and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1994, 3553 RSC.
- A. Mukhopadhyay and S. Pal, J. Chem. Crystallogr., 2005, 35, 737 CrossRef CAS.
- N. Reedig, M. U. Triller, D. Purshe, A. Rompel and B. Krebs, Z. Anorg. Allg. Chem., 2002, 628, 2458 CrossRef.
- D. M. Epstein, S. Choudhary, M. R. Churchill, K. M. Keil, A. V. Eliseev and J. R. Morrow, Inorg. Chem., 2001, 40, 1591 CrossRef CAS PubMed.
- V. Patroniak, A. R. Stefankiewicz, J.-M. Lehn, M. Kubicki and M. Hoffmann, Chemistry, 2006, 144 CAS.
- K. S. Min, T. Weyhermuller, E. Bothe and K. Wieghardt, Inorg. Chem., 2004, 43, 2922 CrossRef CAS PubMed.
- J. Vinsova, V. Horak, V. Buchta and J. Kaustova, Molecules, 2005, 10, 783 CrossRef CAS PubMed.
- J. Jampilek, J. Vinsova and J. Dohnal, 10th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-10), Thesis, http://www.usc.es/congresos/ecsoc/10/ECSOC10.htm%20%26%20http://www.mdpi.org/ecsoc-10/, 1–30 November 2006.
- Y. X. Chen, L. F. Qian, W. Zhang and B. Han, Angew. Chem., 2008, 120, 9470 CrossRef PubMed.
- E. Bayer, Angew. Chem., Int. Ed. Engl., 1964, 3, 325 CrossRef PubMed.
- G. A. Abakumov, V. K. Cherkasov, N. O. Druzhkov, T. N. Kocherova and A. S. Shavyrin, Russ. Chem. Bull., 2011, 60, 112 CrossRef CAS PubMed.
- A. T. T. Hsieh and K. L. Ooi, J. Inorg. Nucl. Chem., 1976, 38, 604 CrossRef CAS.
- S. S. Batsanov, Russ. J. Inorg. Chem., 1991, 36, 3015 CAS.
- A. L. Johnson, N. Hollingsworth, G. Kociok-Kohn and K. C. Molloy, Inorg. Chem., 2008, 47, 9706 CrossRef CAS PubMed.
- I. Biisching and H. Strasdeit, J. Chem. Soc., Chem. Commun., 1994, 2789 RSC.
- V. I. Lodyato, I. L. Yurkova, V. L. Sorokin, O. I. Shadyro, V. I. Dolgopalets and M. A. Kisel, Bioorg. Med. Chem. Lett., 2003, 13, 1179 CrossRef CAS.
- D. D. Perrin, W. L. F. Armarego, D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 1980 Search PubMed.
- G. M. Sheldrick, Structure Determination Software Suite, SHELXTL v.6.12, Bruker AXS, Madison, Wisconsin, USA, 2000 Search PubMed.
- Agilent Technologies, CrysAlis Pro, Agilent Technologies Ltd, Yarnton, England, 2011 Search PubMed.
- G. M. Sheldrick, Bruker/Siemens Area Detector Absorption Correction Program, SADABS v.2.01, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
Footnote |
| † CCDC 772231 and 957572–957574. For crystallographic data in CIF or other electronic format see E-mail: DOI: 10.1039/c3ra47669c |
| This journal is © The Royal Society of Chemistry 2014 |