DOI:
10.1039/C3RA47964A
(Paper)
RSC Adv., 2014,
4, 11528-11534
Switchable fluorescence by click reaction of a novel azidocarbazole dye†
Received
25th December 2013
, Accepted 31st January 2014
First published on 6th February 2014
Abstract
Imaging is – even these days – still restricted to of a few classes of robust dyes. A demand for switchable tags led us to the design of a new class of pre-fluorophores. We achieved this by using a non-fluorescent N-(4-azidophenyl)-carbazole tag which turns fluorescent by click reaction with alkynes and cyclooctynes. The spectral properties of the labelled dyes were investigated. Our results suggest that a twisted internal charge transfer (TICT) transition is responsible for the emission. DFT calculations and single-crystal X-ray diffraction of selected examples support this explanation. The feasibility of the new dyes for biological application has also been tested via confocal microscopy.
Introduction
Carbazole moieties are a structural key motif in both dyes and luminophores. Pigments like Violet 23 or Hidrotin Blue 2R are used e.g. for the staining of clothing and strongly benefit from carbazole units in terms of photostability and high absorbance.1,2 Many fluorescent substances featuring carbazole subunits are known.3–6 A strong, deep blue luminescence is characteristic for most carbazole derivatives, making them broadly used in luminescent devices such as organic light-emitting diodes.7–9 However, because of the π–π* and n–π*-transitions occurring upon excitation of the carbazole core, stokes shifts for carbazole-substituted compounds are usually small. This hampers the discrimination between excitation and emission when using them as dyes in fluorescence microscopy and biological applications.
To resolve this issue, we attempted to design a new fluorescent label, combining the beneficial properties of carbazole such as high photostability and extinction, with the large stokes shift of a charge-transfer (CT) system. CT systems typically consist of a donor-moiety, which may be excited, and a matching acceptor-unit, to which the electron density is shifted. Typically, this leads to broad, ill-structured spectra as well as large stokes shifts. In our case, the carbazole unit acts as a donor-unit, while aryl-substituted 1,2,3-triazoles are used as acceptors (Schemes 1–3).
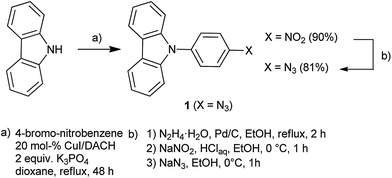 |
| | Scheme 1 Synthetic route to N-(4-azido-phenyl)-carbazole 1. | |
Here, we report the synthesis and the photophysical properties of new carbazole dyes. Single-crystal X-ray diffraction revealed the molecular structure, while DFT- and TD-DFT calculations provided valuable insights into the photophysical mechanism leading to light-emission and a large stokes shift. The dyes themselves are formed by Click-functionalization of a non-fluorescent precursor, N-(4-azidophenyl)-carbazole 1, which is non-fluorescent due to vibronic quenching by the free azido-group. This enables an easy discrimination of labelled material and traces of remaining, non-reacted precursor, e.g. in fluorescence microscopy.
Synthesis of precursor 1 and the fluorescent dyes
The synthesis of the precursor 1 is summarized in Scheme 1. After an Ullmann-type coupling of a straight carbazole with 4-bromo-nitrobenzene, the resulting N-(4-nitrophenyl)-carbazole is reduced with hydrazine, diazotized with HNO2 and converted to N-(4-azidophenyl)-carbazole 1 by reaction with sodium azide. Further functionalization of non-fluorescent N-(4-azidophenyl)-carbazole 1 with terminal alkynes in a Cu(I)-catalysed alkyne–azide cycloaddition (CuAAC) (Scheme 2) as well as with cyclooctynes (Scheme 3) yielded the fluorescent carbazole dyes.10,11
 |
| | Scheme 2 Reaction of N-(4-azido-phenyl)-carbazole 1 with terminal alkynes. | |
 |
| | Scheme 3 Reaction of N-(4-azidophenyl)-carbazole 1 with cyclooctynes. | |
For the CuAAC click reaction, we chose copper(I) iodide in N,N-diisopropylethylamine (DIPEA) as the catalyst and several alkyne moieties such as the commercially available phenylacetylene as well as 2-methyl-3-butyn-2-ol, yielding the fluorescent dyes 2 and 3, respectively. High conversion was achieved for both examples (92% yield each) within 4 hours. Furthermore, azide 1 was coupled to a polycationic peptoid containing an alkyne side chain on solid phase in a submonomer approach under the same conditions (see ESI†).12,13 This peptoid is a heptameric compound with six aminohexyl- and one propargyl-side chain and was used to investigate the influence of charged moieties on the carbazol fluorescence.14,15
In contrast to the CuAAC reaction, the reaction between azides and cyclooctynes does not require any catalysts at room temperature due to the strained ring structure of cyclic alkyne. This makes the strain-promoted azide–alkyne cycloaddition (SPAAC)16 suitable for click reactions in the presence of cells or whole organisms. All reactions depicted in Scheme 3 were carried out by simply dissolving the reactants (azide 1 and one equivalent of cyclooctynes 5a–8a) in dichloromethane (DCM) and stirring overnight to yield dyes 5–8 in good to very high yields (74–98%).
Single-crystal X-ray diffraction
We were able to characterize molecules 2 and 3 by single-crystal X-ray diffraction (Fig. 1 and ESI†).17 The most striking observation for both structures was the twisting of all three π-systems, which breaks the conjugation between the three ring systems into electronically independent localized moieties. The smallest angle between the triazole ring and the phenyl ring is 29.9° for component 2 and 38.7° for component 3. Between the carbazole moiety and the phenyl ring, an angle of 49.4° for component 2 and 66.2° for component 3 has been found. The molecular structures of 2 and 3 have also been used as starting structures of DFT-calculations and to interpret the photophysical properties (see below).
 |
| | Fig. 1 Molecular structure of 3 (displacement parameters are drawn at 50% probability level). | |
Absorption spectroscopy
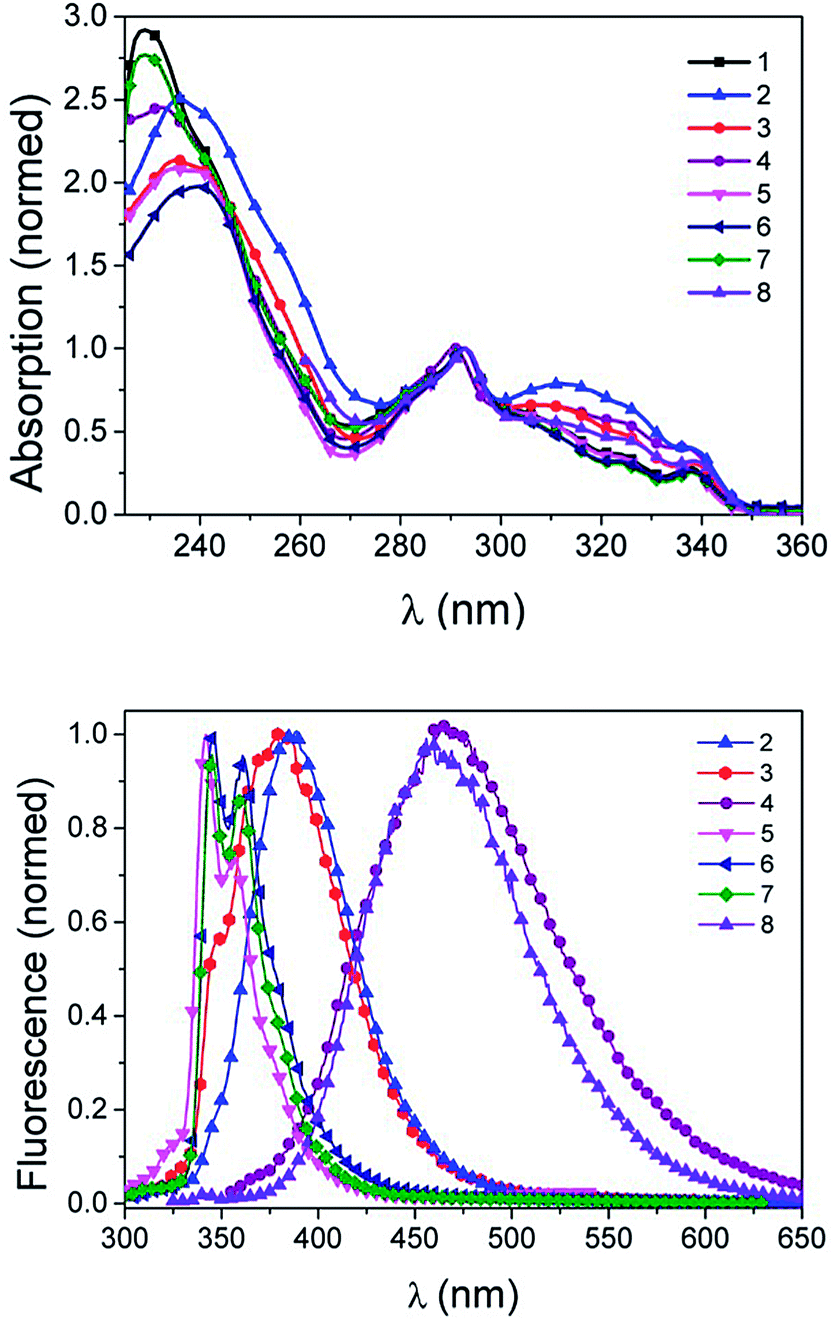
It is tempting, yet illegitimate to interpret a twist of the π-systems in solution from the results found in single-crystals without further evidence. We thus recorded UV-Vis spectra in various solvents. In summary, absorption spectra of both the unreacted precursor 1 and the fluorescent compounds 2 to 8 were quite similar. Carbazole moieties are very strong chromophores, which dominate the UV-Vis spectra; Peaks varied only in a 4 nm range (Fig. 2, Table 1 and ESI†). Molar absorption coefficients were quite high and depended on the solvents (Table 1 and ESI†). Substitutions with aromatic rings and modifications of side groups did not lead to a bathochromic shift or strong changes in the absorption spectra (Fig. 2 and Table 1). This suggested that the twisting of the conjugation found in solid state by X-ray diffraction also occurred in solution. Subtle variations manifested themselves in the differences in height of the shoulders in the normalized absorption spectra. Due to the broken conjugation, the UV-Vis spectra may be described as a superposition of the expected spectra of the isolated chromophores (phenyl, triazol, carbazole) with additional bands due to a charge-transfer transition from the carbazole to the aryl-triazole (300–350 nm).
 |
| | Fig. 2 Absorption (top) and emission (bottom) spectra of compounds 1–7 in DCM, 8 in DMSO. Excitation wavelength λex = 290 nm. | |
Table 1 Spectroscopic data for the compounds at room temperature
| |
λ
abs (nm) |
ε (M−1 cm−1) |
λ
em (nm) |
Φ
|
Stokes shift |
|
Solid,
DCM,
DMF,
H2O,
MeOH,
DMSO,
Cyclohexane,
Diethyl ether,
Ethyl acetate,
DMF.
|
|
1
b
|
293 |
21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 ± 600 300 ± 600 |
|
|
|
|
2
b
|
292 |
2400 ± 2000 |
387 |
0.72 ± 0.051 |
95 |
|
3
a
|
292 |
|
492 |
0.41 ± 0.03 |
200 |
|
3
b
|
292 |
15700 ± 500 |
384 |
0.365 ± 0.030 |
92 |
|
3
c
|
292 |
23500 ± 800 |
362 |
0.468 ± 0.046 |
70 |
|
3
e
|
291 |
19800 ± 700 |
424 |
|
132 |
|
4
a
|
291 |
|
434 |
|
143 |
|
4
d
|
291 |
4000 ± 200 |
467 |
0.027 ± 0.001 |
176 |
|
4
e
|
291 |
3800 ± 250 |
431 |
0.102 ± 0.005 |
140 |
|
5
a
|
293 |
|
415 |
|
122 |
|
5
b
|
293 |
1950 ± 50 |
342/356 |
0.311 ± 0.081 |
49/63 |
|
6
a
|
293 |
|
490 |
|
197 |
|
6
b
|
293 |
19600 ± 900 |
345/360 |
0.319 ± 0.040 |
52/67 |
|
7
a
|
293 |
|
492 |
|
199 |
|
7
b
|
293 |
12400 ± 300 |
345/360 |
0.284 ± 0.058 |
52/67 |
|
7
c
|
291 |
32000 ± 7000 |
345/362 |
0.317 ± 0.036 |
54/71 |
|
7
e
|
291 |
11100 ± 1000 |
431 |
0.347 ± 0.049 |
140 |
|
7
g
|
292 |
|
341/357 |
|
49/65 |
|
7
h
|
291 |
|
342/357 |
|
51/66 |
|
7
i
|
291 |
|
344/360 |
|
53/69 |
|
7
j
|
291 |
|
345/362 |
|
54/71 |
|
8
a
|
293 |
|
416 |
|
123 |
|
8
f
|
293 |
28000 ± 3000 |
459 |
0.552 ± 0.078 |
166 |
Emission spectroscopy
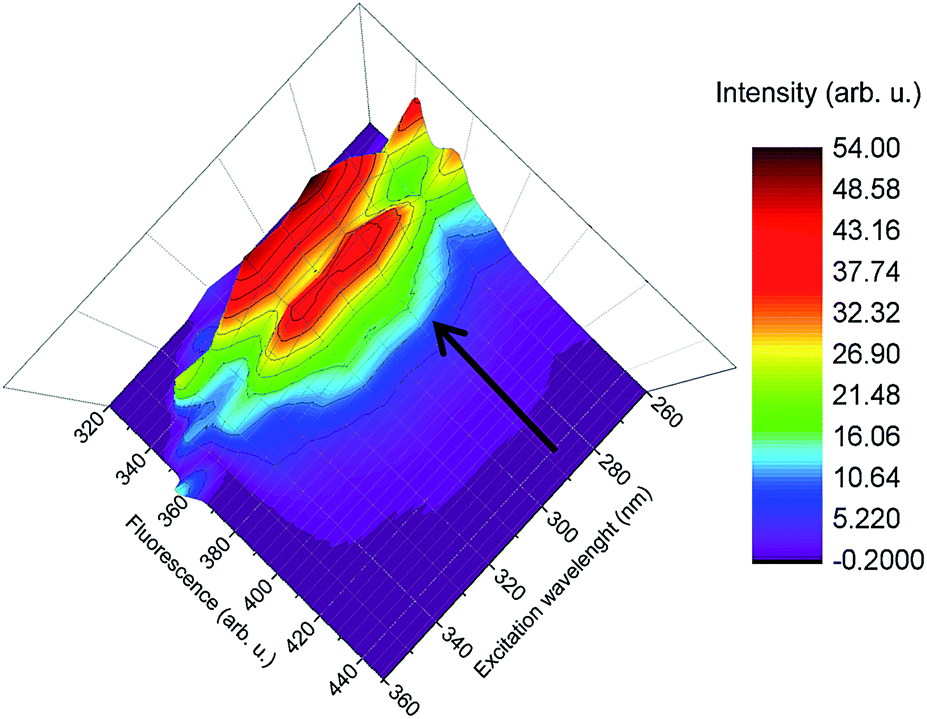
To evaluate the strongest transition between absorption and emission 3D spectra of compound 7 in ethyl acetate and cyclohexane have been recorded. The result for cyclohexane is given in Fig. 3, while the corresponding plot for ethyl acetate is given in the ESI.† For both solvents, 3D-spectra were similar and in agreement with the 2D-measurements (fluorescence over wavelength, see Fig. 2) over a range of excitation wavelengths. The strongest transition was found to occur at an excitation wavelength of 292 ± 1 nm. Excitation with lower energies (345 nm) did not lead to detectable luminescence. We thus define the strong transition at 292 ± 1 nm as the preferred excitation wavelength and refer the stokes shifts to this transition.
 |
| | Fig. 3 3D fluorescence spectra of compound 7 in cyclohexane. λex= 260–370 nm, excitation steps in 10 nm difference, λem= 320–450 nm. | |
Solvent- and polarity-influences on the emission
Emission spectra of the novel carbazole dyes were highly dependent on the solvent, with varying emission maxima between 341 nm and 492 nm (Table 2, Fig. 4).
Table 2 Solvent dependency for compound 7 in numbersa
| Solvent |
Orientation polarizability (Δf) |
Stokes shifts [nm] |
Stokes shift 1 [104 cm−1] |
Stokes shift 2 [104 cm−1] |
Mean [104 cm−1] |
|
DCM = dichloromethane, DMF = dimethylformamide; calculation of stokes shifts see ESI. Δf numbers for methanol and ethyl acetate, as well as formula according to ref. 18.
|
| Cyclohexane |
0 |
49/65 |
0.49 |
0.65 |
0.57 |
| Diethyl ether |
0.17 |
51/66 |
0.51 |
0.66 |
0.585 |
| Ethyl acetate |
0.201 |
53/69 |
0.53 |
0.69 |
0.61 |
| DCM |
0.217 |
52/67 |
0.52 |
0.67 |
0.595 |
| DMF |
0.274 |
54/71 |
0.54 |
0.71 |
0.625 |
| Methanol |
0.310 |
140 |
1.40 |
|
1.40 |
 |
| | Fig. 4 Lippert–Mataga-plot of compound 7. | |
However, differences between compounds 2–4 and 5–7 were noted probably due to structural variations. While compounds 2–4 have been reacted with terminal alkynes, compounds 5–8 have been reacted with cyclooctynes and contain bicyclic (5–7) and polycyclic (8) moieties. Stokes shifts were large, ranging from 49 nm to 200 nm and strongly solvent dependent (see Tables 1 and 2). The Lippert–Mataga-Plot, which was plotted for substance 7, also served to rationalize this (see Fig. 4). A linear correlation of the solvent orientation polarisability and the obtained stokes shift was found (slope of the line and standard deviation in ESI†). The orientation polarisability of the environment interacted with the dipole moment of the chromophore. This led to a shift of the emission maxima and consequently to the large stokes shifts.
For methanol (MeOH), we noticed significant deviations from the fitted line in the Lippert–Mataga plot. Interestingly, no such effects were found in absorption spectra (Fig. 5). We recorded emission spectra of compound 7 in different mixtures of MeOH and DCM as solvents. In non-protic solvents such as DCM, two small band maxima were found, while an additional, broad band appeared when protic methanol was mixed with the non-protic solvent. When increasing the amount of protic solvent, the broad band became more pronounced.
 |
| | Fig. 5 Absorption (top) and emission (bottom) spectra of compound 7 in solvent mixtures of MeOH and DCM. Excitation wavelength λex = 290 nm. | |
This behaviour is indicative for the occurrence of different emissive states. The blue-shifted, structured transition could be attributed to carbazole-centered fluorescence, which was apparently not influenced by the surrounding solvents. The second, red-shifted transition was stabilized by protic solvents and was thus more pronounced when moving from pure DCM to pure MeOH. Under aqueous conditions, the carbazole-based emission was fully suppressed in favour of the CT-type band as shown for the water soluble compound 4. The fact that increasing polarity and the presence of protic solvents seem to cause a very large stokes shifts makes these dyes interesting candidates for biological applications.
pH-influences on the emission for compound 4
Compound 4 carried six amino groups and was therefore expected to be influenced by the pH value of the medium due to the introduction of positive charges in close proximity to the emissive chromophore. The absorption and emission spectra have been measured in phosphate buffered saline (PBS) under acidic, neutral and basic conditions (spectra see Fig. 6 and ESI†). The emission spectra (Fig. 6) were pH sensitive: The band was shifted towards lower energies, when moving to more acidic solvents. This was accompanied by decreased quantum efficiency. Similar to the influences of protic solvents on compound 7, the pH-value did not seem to affect the absorption spectra of compound 4. We thus presume that the positive charge of the ammonium groups influenced the molecules in excited state, which is typical for CT emission. This decreased the intensity and shifted the emission to longer wavelength (normalized spectra see ESI†).
 |
| | Fig. 6 Fluorescence spectra of compound 4 in different pH, the intensities are recalculated to the lamp intensities and detection sensitivity. Excitation wavelength λex = 290 nm. | |
Determination of the quantum efficiency and emission decay time
Also measurements of the photoluminescence quantum yield (PLQY)19 were done in different solvents as well as in the solid state (Table 1). The PLQY values were quite high, despite the large stokes shift. They mostly varied between 30% and 72%. An exception was compound 4 due to the high interactions with the protonated amino side chains, as stated above (ESI†). Using the time-correlated single photon counting (TCSPC) technique, the emission decay time of components 2 and 3 was measured in degased DCM at room temperature. Both compounds featured a monoexponential decay with τ = 3 and 4 ns, respectively. These lifetimes indicated a fluorescence emission without an involvement of triplet state (T1). We can exclude delayed fluorescence as the cause for the broad spectra and large stokes shifts in the solid state.
DFT calculations
Density functional theory (DFT) geometry calculations for compounds 2, 3 and 7 were performed on B3-LYP/def2-TZVP level20,21 using D3 dispersion corrections.22 Comparison of the calculated and measured bond lengths and angles (see ESI†) showed a good agreement for the applied functional. Representative frontier orbitals (HOMO and LUMO) for compound 3 are depicted in Fig. 7 (see ESI† for compounds 2 and 7). For all calculated compounds, the highest occupied molecular orbital (HOMO) is mainly localized at the carbazole moiety, whereas the lowest unoccupied molecular orbital (LUMO) is localized on the triazole and bridging phenyl ring. As seen for the ground state HOMO–LUMO energies in Table 3, HOMO energies remain basically unchanged (around −5.7 eV) with varying substituents at the triazole, whereas the LUMO energies are affected. Hence, it appears that major differences in the observed emission maxima of compounds 2, 3 and 7 may result from variations in the LUMO energies.
 |
| | Fig. 7 HOMO (left) and LUMO (right) frontier orbital plots of compound 3. | |
On the basis of these results, the type of electronic transition that led to the lowest excited states may be classified as being largely based on intramolecular charge-transfer character. This assumption was further supplied by gas phase time-dependent density functional theory (TD-DFT) calculations at the same level, which shows 99% participation of HOMO → LUMO for the S0 to S1 vertical singlet excitation. However, it is known that exact DFT calculations of charge-transfer (CT) excitations are very difficult. Since all excitations show a distinct CT character, the excited states and transition energies are not described quantitatively by simple application of TD-DFT. As found for compound 3 (Table 3), the energies of HOMO and LUMO are just slightly affected by varying solvent polarity (conductor-like screening model, COSMO).23
Table 3 B3LYP/def2-TZVP HOMO and LUMO energies (in eV) at the ground state, left: of 2, 3, 7 (gas phase); right: of 3 with different solvents (COSMO)
| |
2
|
3
|
7
|
3 (CH2Cl2) |
3 (MeOH) |
| HOMO |
−5.72 |
−5.71 |
−5.76 |
−5.70 |
−5.70 |
| LUMO |
−1.62 |
−1.52 |
−1.38 |
−1.42 |
−1.41 |
Twisted internal charge transfer mechanism
All results indicates that a twisted internal charge transfer (TICT) as described by Wang et al.24 is responsible for the emission of our new dyes. We believe that the strong polarity dependence of the stokes shift originates from solvent dependent slow reorientations of the molecule dipole in the excited states. This is in good agreement with all photophysical measurements, the TCSPC measurements, X-ray results and DFT calculations. The reason for this is the polarity-dependent twist of the three connected π-systems of the carbazole, phenyl and triazole moieties.
Biological application
To verify that the specific conditions in biological media lead to a twisting conformation that allows for efficient light detection, we tested compounds 2 and 7 for their applicability in cell culture. Eventually, 104 human cervix carcinoma (HeLa) cells were treated with 30 μM of 2 and 7, respectively.
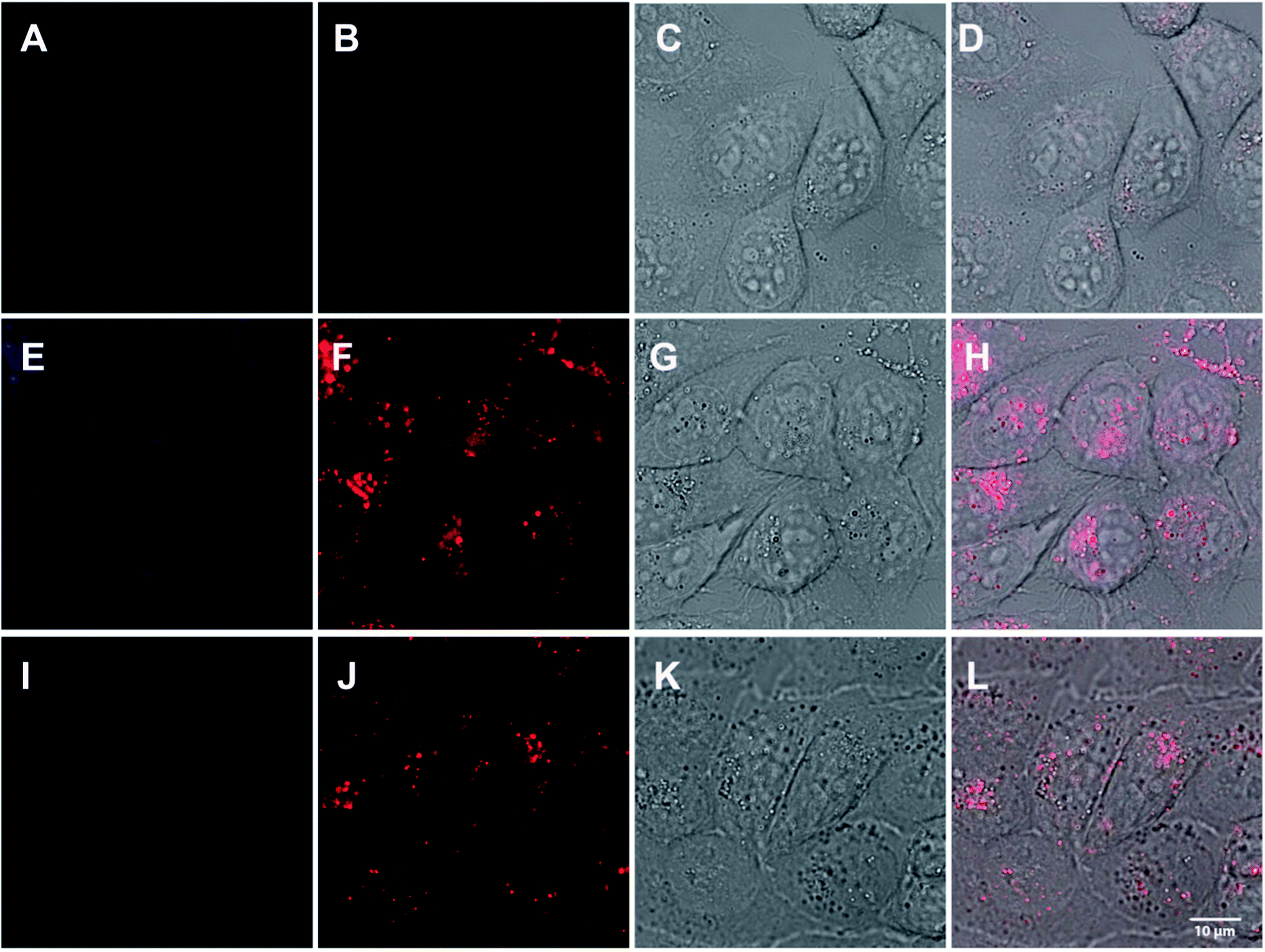
Fluorescence confocal microscopy showed that in cells both compounds revealed a strong fluorescence with a bathochromic shift of the measured emission maxima towards 450–550 nm, when excited with a UV laser at 351 nm. Both compounds 2 and 7 showed a strong accumulation within the endosomal-lysosomal system (Fig. 8), which suggests an endocytotic uptake. A xyλ-scan resulted in maximum fluorescence emission at about 481 nm for 2 and 543 nm for 7. While the bathochromic shift of compound 2 is less pronounced, the shift in 7 is in accordance with the fluorescence measurements in protic solvents with increasing polarity (Fig. 5).
 |
| | Fig. 8 104 HeLa cells were incubated with 30 μM of 2 (E–H) or 7 (I–L) in DMEM (20 mM stock solution in DMSO, freshly diluted 666 × in DMEM) at 37 °C for 24 h and subsequently subjected to fluorescent confocal microscopy using a Leica SP5-TCS (DMI6000) inverse microscope. Objective: HCX PL APO CS 63.0 × 1.20 WATER UV. Excitation wavelength: 351 nm. 1st column (A, E and I): Emission bandwidth: 417–468 nm, 2nd column (B, F and J): emission bandwidth: 518–724 nm, 3rd column (C, G and K): Brightfield, 4th column (D, H and L): Merge. A–D: Control cells, only DMSO, E–H: Compound 2, I–L: Compound 7. | |
As the pH of the endocytic pathway is decreasing from the plasmamembrane towards the lysosomal compartment (from 7.4 to 4.4), the red-shifted transition of compound 7 is shifted towards lower energies when moving to more acidic (and thus, more protic) compartments. This is even more pronounced in cells than in solution. Although the excitation wavelength is not optimal, the strong fluorescence emission of the compound promises an application in biological systems.
Conclusions
In this study, we present a new class of fluorescent carbazole dyes. We demonstrate that the non-fluorescent N-(4-azidophenyl)-carbazole moiety becomes fluorescent by click reaction with an alkyne or cyclooctyne. This specific property allows fluorescent control in samples after click reaction, for example in biological samples or material science. The spectral properties show that absorption spectra are not changed after click reaction with various substrates. The fluorescence of the resulting dyes results from a twisted internal charge transfer mechanism, which has been verified by detailed photophysical experiments, single-crystal X-ray diffraction and DFT-calculations. The leads to a high solvent dependency accompanied by large stokes shifts from 49–200 nm. Most samples have a high quantum yield between 30 and 72%. The feasibility of the new dyes has been demonstrated in a first series of confocal microscopy experiments.
Acknowledgements
We acknowledge the Carl-Zeiss-Stiftung (A. H. and D. F.), Karlsruhe School of Optics and Photonics (KSOP) (A. H. and D. K. K.) and the Landesgraduiertenförderung Baden-Württemberg (T. H. and L. G.) for and financial support. We thank cynora GmbH for support. The work was further supported by the Helmholtz programme Biointerface (U. S. and S. B.). We also acknowledge the working group of Dr L. Fruk at the DFG-Center for Functional Nanostructures, Karlsruhe Institute of Technology for using their infrastructure.
Notes and references
- Y. F. Lan and J. J. Lin, Dyes Pigm., 2011, 90, 21–27 CrossRef CAS PubMed.
- R. Levinson, P. Berdahl and H. Akbari, Sol. Energy Mater. Sol. Cells, 2005, 89, 351–389 CrossRef CAS PubMed.
- W. Maes, T. H. Ngo, G. Rong, A. S. Starukhin, M. M. Kruk and W. Dehaen, Eur. J. Org. Chem., 2010, 2576–2586 CrossRef CAS.
- M. Tavasli, T. N. Moore, Y. H. Zheng, M. R. Bryce, M. A. Fox, G. C. Griffiths, V. Jankus, H. A. Al-Attar and A. P. Monkman, J. Mater. Chem., 2012, 22, 6419–6428 RSC.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- G. Moad, M. Chen, M. Haussler, A. Postma, E. Rizzardo and S. H. Thang, Polym. Chem., 2011, 2, 492–519 RSC.
- S. Y. Takizawa, V. A. Montes and P. Anzenbacher, Chem. Mater., 2009, 21, 2452–2458 CrossRef CAS.
- Y. Heischkel and H. W. Schmidt, Macromol. Chem. Phys., 1998, 199, 869–880 CrossRef CAS.
- D. Volz, T. Baumann, H. Flügge, M. Mydlak, T. Grab, M. Bächle, C. Barner-Kowollik and S. Bräse, J. Mater. Chem., 2012, 22, 20786–20790 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- H. Jang, A. Fafarman, J. M. Holub and K. Kirshenbaum, Org. Lett., 2005, 7, 1951–1954 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- D. K. Kölmel, D. Fürniss, S. Susanto, A. Lauer, C. Grabher, S. Bräe and U. Schepers, Pharmaceuticals, 2012, 5, 1265–1281 CrossRef PubMed.
- T. Schröder, K. Schmitz, N. Niemeier, T. S. Balaban, H. F. Krug, U. Schepers and S. Bräse, Bioconjugate Chem., 2007, 18, 342–354 CrossRef PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- Structure solution and refinement program SHELXL-97: G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 2nd edn., 1999, p. 189, table 6.2 Search PubMed.
- S. Fery-Forgues and D. Lavabre, J. Chem. Educ., 1999, 76, 1260–1264 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- J. Wang, Y. Wang, T. Taniguchi, S. Yamaguchi and S. Irle, J. Phys. Chem. A, 2012, 116, 1151–1158 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterisation, spectra, computation method and X-ray crystallographic information files for compounds 2 and 3 (CIF). CCDC 936576 and 936577. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47964a |
| ‡ Both authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a