DOI:
10.1039/C3RA47922F
(Paper)
RSC Adv., 2014,
4, 10317-10329
Utilizing thiol–ene coupling kinetics in the design of renewable thermoset resins based on D-limonene and polyfunctional thiols†
Received
23rd December 2013
, Accepted 31st January 2014
First published on 3rd February 2014
1. Introduction
The search for valuable compounds from renewable resources to prepare non-petroleum plastic materials has grown into an important topic in the polymer field not only on grounds of increasing environmental awareness and issues of sustainable development but also because fossil-oil reserves are dwindling at an increasingly faster rate. The development of reaction systems that meet the demands of ‘green chemistry’ with low energy requirements and eco-friendly synthesis routes, just to mention a few, are also becoming a matter of increasing interest. An extensive array of starting materials comprised of vegetable oils, lignin, polysaccharides (e.g., cellulose and starch), sugars/carbohydrates, polycarboxylic acids, glycerol, macrocyclic lactones, furans, etc., have all been explored either as they are found natively or modified to prepare bio-based polymers and biocomposites using a wide variety of chemistries and enzymatic methods.1,2 The use of natural olefins, most of which are biodegradable, have also been studied3 and their combination with environmentally friendly and economically favorable UV technology offers a viable ‘green’ alternative to petrochemical-based monomers and resulting plastics and complies well with regulations within the coating Industry.
Macromolecular ultraviolet (UV)-curing thiol–ene systems are also acquiring high popularity in industry and scientific research community due to industrially favoring characteristics such as: resistance to inhibition by ambient oxygen (acting as chain stopper) which is a serious problem affecting conventional acrylic-based chain-growth radical polymerizations; relatively rapid cure rates leading to highly cross-linked networks; solventless processing when thiol and ene co-reactants are miscible; improved photocuring control (both spatial and temporal); ability to initiate polymerization without addition of photoinitiator thus enabling the cure of thick geometries; and, a step-wise radical growth mechanism leading to delayed gel-point which results in thermosets with uniform cross-link density, narrow glass-transition temperatures, reduced volume shrinkage and low stress development at high monomer conversions.4–6 Additionally, thiol–ene photopolymers are transparent and exhibit enhanced physical properties such as mechanical flexibility and good adhesion to several substrates.6,7 This makes thiol–ene photopolymers ideal for organic coating applications.
A group of renewable monomers with significant potential in this context is terpenes. A very common monoterpene, D-limonene, is present in large amounts in the rind of oranges and related fruits and thus can be obtained as by-product of the citrus industry.8 This chiral compound retains in its chemical structure two unconjugated electron-rich alkenes with different degrees of substitution susceptible for radical polymerization: a terminal 1,1-disubstituted vinylidene (or isopropenyl) functionality and an internal 1,1,2-trisubstituted 1-methyl-cyclohexene moiety. Furthermore, the slightly rigid cycloaliphatic ring may help introduce some degree of mechanical integrity to conventional thiol–ene networks as opposed to their homologous acrylates.9–11 One striking feature of using limonene in photopolymerization systems is the possibility to obtain novel tridimensional network structures as result of the multi-chirality introduced by the monoterpene upon C–S bond formation via thiol–ene coupling with polyfunctional thiols.12
Thiol–ene chemistry, although already known for more than a century, has not been extensively used in the formation of thermosets derived from renewable alkene monomers and very little is actually known about the thermo-mechanical properties of the ensuing polymeric materials. For instance, epoxidized linseed oil (ELO) was thermally reacted with grapeseed oil (GSO) pre-functionalized with cysteamine chloride via UV-induced thiol–ene coupling to create a cross-linked material based on two bio-renewable sources of vegetable oils.13 Also, several allyl-, acrylate-, and vinyl-ether derivatives of the triglyceride ricinolein (the chief unsaturated constituent of castor oil) have been synthesized and combined with multifunctional thiols to prepare UV-cured thiol–ene networks. Testing on the film-materials immediately after UV-exposure and one week later indicated increased cross-linking density and superior physical properties upon aging.14
Conventionally, the thiol–ene coupling reaction proceeds as a typical free-radical chain process with initiation, propagation–chain-transfer and termination steps. In a previous paper15 we have examined the kinetics of photo-catalyzed free-radical thiol–ene additions involving D-limonene and two mono-/tri-functional alkyl ester 3-mercaptopropionates to understand the correlation between alkene structure and reactivity towards this particular class of thiol. We found that thiol–ene coupling at the exo-olefinic bond proceeds about 6.5 times faster than at the endocyclic one in organic solution. This value for the regioselectivity was attributed primarily to a higher relative energy of the tertiary β-thioether carbon-radical intermediate resulting from thiyl-radical insertion across the trisubstituted double-bond; and moderately, to steric impediments controlling thiyl radical addition onto the two distinct unsaturations. These two combined effects account for the relatively low conversion rates observed for the two multi-substituted enes and internal cis/trans-1,2-disubstituted olefins,16,17 when compared to more reactive enes (e.g., vinyl ethers)6,18 owed to highly reversible propagation steps in addition to a chain-transfer rate-limiting step that controls the overall reaction.
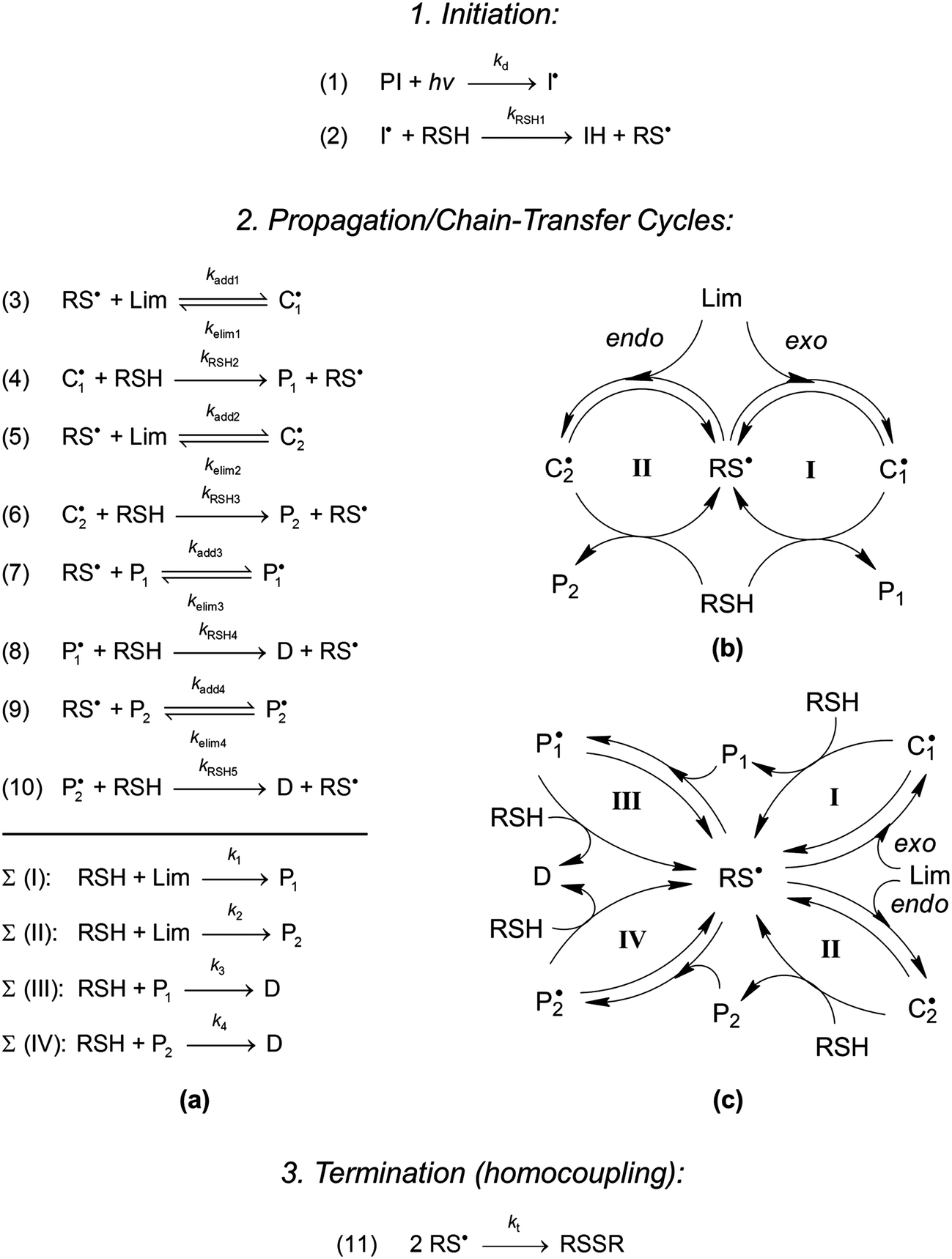
In this paper, we wish to develop further this concept by exploring the influence of the reaction conditions in bulk, namely the method of initiation (thermal versus photoinduced) as well as the kinetic effect of initial co-reactant stoichiometry, on the overall thiol–ene coupling dynamics. Our goal is to understand which of these operational parameters is more significant for the synthesis of functional terpene-based resins and resulting polymeric materials. Specifically, we propose an extension to the fundamental two-route linear cyclic mechanism depicted in Scheme 1b describing the basic step-wise reaction sequence that governs the selective thiol–ene coupling kinetics in D-limonene15 and how manipulating the starting composition stoichiometry impacts the kinetic behavior and outcome of the reaction. The underlying mechanism detailed in Scheme 1a/c, with initiation and termination reactions, was tested numerically via computer simulation by comparison with experimental kinetic data. The resulting model investigations aim to complement a previous study15 while providing a fundamental kinetic basis towards the development of new cross-linkable poly(thioether) networks based on limonene.
 |
| | Scheme 1 Suggested mechanistic scheme for the step-growth thiol–ene coupling reaction: (a) sequence of elementary propagation–chain-transfer steps and set of overall coupling reactions resulting from the free-radical thiol–ene additions involving the two unsaturations of D-limonene accounting for primary and secondary coupling; (b) graphical representation of the basic two-route linear mechanism (steps (3)–(6) and observed global reactions (I)–(II)) with three transitory radical species sharing a common intermediate radical (RS˙); and, (c) complex multi-route linear mechanism with five transitory radical species having one common intermediate (RS˙) connecting the four thiol–ene coupling cycles. At steady-state the concentration of intermediate radicals are assumed to remain constant. | |
2. Experimental section
2.1. Materials
(R)-(+)-limonene (Lim 1, ≥ 97%) was obtained from Sigma-Aldrich (Sweden). The thiols iso-tridecyl 3-mercaptopropionate (C13MP, 2) and trimethylolpropane tris(3-mercaptopropionate) (TMPMP, 6) were kindly provided by Bruno Bock Chemische Fabrik GmbH & Co (Marschacht, Germany). The photoinitiator, 2,2-dimethoxy-2-phenylaceto-phenone (Irgacure 651/DMPA), was obtained from Ciba Specialty Chemicals Inc. (Switzerland). Thermal initiator 2,2′-azobis(2-methylpropionitrile) (AIBN) was acquired from Fluka. Ethyl acetate (EtOAc, 99%) was obtained from Merck (Darmstadt, Germany). Deuterated chloroform (CDCl3, 99.8%) was provided by CIL (Cambridge Isotope Laboratories, Inc., USA). All reagents and solvents were obtained at the highest purity available from the above commercial companies and used as received without further purification.
2.2. Instrumentation
2.2.1. UV-light sources. A Hamamatsu L5662 equipped with standard medium pressure 200 W L6722-01 Hg–Xe lamp provided with optical fibers was used as the UV-source for the discontinuous kinetic studies. The UV-light intensity delivered was measured using a Hamamatsu UV-light power meter (model C6080-03) calibrated for the main emission line centered at λ = 365 nm. The light source used for resin synthesis was a Blak Ray B-100AP (100 W, λ = 365 nm) Hg UV-lamp with an irradiance of ∼25 mW cm−2 as determined with an UVICURE Plus High Energy UV Integrating Radiometer (EIT, USA), measuring UVA at 320 ≤ λ ≤ 390 nm.
2.2.2. NMR Spectroscopy. 1H NMR spectra of the samples were recorded on a 400 MHz Bruker Aspect NMR spectrometer (Karlsruhe, Germany). 1H NMR spectra (128 scans) were acquired with a spectral window of 20 ppm, an acquisition time of 4 seconds, and a relaxation delay of 2 seconds. Analytes were prepared by dissolving 8.0 mg of sample in 0.8 ml of deuterated chloroform (CDCl3) containing 0.05% of TMS in a 5.0 mm diameter glass tube. Chemical shifts (δ) were reported in parts per million (ppm) relative either to the tetramethylsilane (TMS) reference signal at 0.00 ppm or residual non-deuterated solvent (CHCl3) signal located at 7.26 ppm. Spectral analysis was performed using the Mestrec software.
2.2.3. FT-Raman spectroscopy. Fourier Transform Raman Spectroscopy spectra were acquired for all samples using a Perkin-Elmer Spectra 2000 NIR-Raman instrument with Spectrum software. Each spectrum collected was based on 32 scans using a 103 mW laser power.
2.2.4. DMF–SEC analyses. Size-exclusion chromatography (SEC) measurements were performed on a TOSOH EcoSEC HLC-8320GPC system equipped with an EcoSEC RI detector and three columns (PSS PFG 5 μm; Microguard, 100 Å and 300 Å) (MW resolving range: 300–1.0 × 105 Da) from PSS GmbH. Measurements were carried out at 50 °C using DMF solvent supplemented with 0.01 M LiBr as mobile phase (isocratic elution, 0.2 ml min−1). A conventional calibration method was created using narrow linear poly(methyl methacrylate) standards. Corrections for flow rate fluctuations were made using toluene as an internal standard. PSS WinGPC Unity software version 7.2 was used to process and analyze the data. Analytes were dissolved in DMF/LiBr solvent (2.5 mg ml−1) and the resulting solutions filtered with a 0.45 μl teflon filter before analysis.
2.3. Procedures
2.3.1. Sample preparation and reactivity assessment. For model studies with C13MP, several reaction mixtures differing in ratio of thiol and alkene functionalities were prepared in bulk conditions in the presence of a small amount of initiator (2.0 wt%). The amount of thiol-to-ene equivalents was adjusted to a mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:0.5 (i.e., 2:1 and 1:1 if expressed in terms of thiol–terpene molecular ratio, respectively). For thermally induced reactions, a mother solution without initiator was first prepared in a 250 ml round-bottom flask. The mixture was immediately degassed in ice-cold bath under reduced pressure and magnetic stirring for 45 min and then purged with dry argon gas for 25 min to flush-out residual dissolved oxygen and restitute the headspace with an inert atmosphere. Small-scale deaerated flat-bottom glass tubes of cylindrical shape (2.5 cm diameter, 10 cm height) already containing the right amount of AIBN were then charged with 8.0 ml of the monophasic liquid and the resulting mixture allowed to react in inert atmosphere. The reactions were conducted in triplicate as closed-systems under magnetic agitation (300 rpm) at a temperature maintained between 68 to 70 °C. Aliquot samples (1.0 ml) were taken periodically at times t = 0, 6, 24, 48 and 120 hours, and then stored at −20 °C until further use. For the photo-initiated reactions, a volume of 2.0 ml from each of the two different stoichiometric mixtures was transferred to small cylindrical glass vessels (40 × 12 mm) used as mini-reactors. The individual samples were UV-irradiated (∼4.2 mW cm−2) for a given time interval at room-temperature and in the presence of ambient air under continuous magnetic stirring kept at 103 rpm. Organic synthesis of the multifunctional resins based on D-limonene (1) and TMPMP (6) were performed photochemically using two different stoichiometric compositions: in the first formulation the two co-reactants were mixed on a half-mole alkene functional group stoichiometry with respect to the thiol and then dissolving the two monomers in 50 wt% of ethyl acetate (EtOAc) with respect to the total weight of reactants. In the second formulation, a 10-fold mole excess of limonene in regard to the thiol functionality was introduced. Both samples were supplemented with a small catalytic amount of DMPA (∼1.0 wt% of total mixture) to accelerate the coupling reaction. The two distinct binary mixtures were transferred to cylindrical glass beakers and exposed to UV-irradiation delivering a constant light irradiance of 25 mW cm−2 (λmax = 365 nm) for a period of 6 hours which corresponds to an overall accumulated dose of 540 J cm−2. Variation in the incident UV-light intensity was observed not to exceed the 6.0% in the radial direction measured at the liquid surface. The photo-induced reactions were conducted at room temperature in the presence of ambient air under magnetic stirring (∼800 rpm). Diluting solvent was removed via rotor-evaporation at 50 °C followed by normal vacuum until residual ethyl acetate was eliminated. Resultant crude synthesis products were obtained as clear, slightly yellow, viscous liquids exhibiting a fresh citreous odor. All mixtures were prepared immediately prior to each experiment in order to ensure reproducibility of sample history.
:1 and 1:0.5 (i.e., 2:1 and 1:1 if expressed in terms of thiol–terpene molecular ratio, respectively). For thermally induced reactions, a mother solution without initiator was first prepared in a 250 ml round-bottom flask. The mixture was immediately degassed in ice-cold bath under reduced pressure and magnetic stirring for 45 min and then purged with dry argon gas for 25 min to flush-out residual dissolved oxygen and restitute the headspace with an inert atmosphere. Small-scale deaerated flat-bottom glass tubes of cylindrical shape (2.5 cm diameter, 10 cm height) already containing the right amount of AIBN were then charged with 8.0 ml of the monophasic liquid and the resulting mixture allowed to react in inert atmosphere. The reactions were conducted in triplicate as closed-systems under magnetic agitation (300 rpm) at a temperature maintained between 68 to 70 °C. Aliquot samples (1.0 ml) were taken periodically at times t = 0, 6, 24, 48 and 120 hours, and then stored at −20 °C until further use. For the photo-initiated reactions, a volume of 2.0 ml from each of the two different stoichiometric mixtures was transferred to small cylindrical glass vessels (40 × 12 mm) used as mini-reactors. The individual samples were UV-irradiated (∼4.2 mW cm−2) for a given time interval at room-temperature and in the presence of ambient air under continuous magnetic stirring kept at 103 rpm. Organic synthesis of the multifunctional resins based on D-limonene (1) and TMPMP (6) were performed photochemically using two different stoichiometric compositions: in the first formulation the two co-reactants were mixed on a half-mole alkene functional group stoichiometry with respect to the thiol and then dissolving the two monomers in 50 wt% of ethyl acetate (EtOAc) with respect to the total weight of reactants. In the second formulation, a 10-fold mole excess of limonene in regard to the thiol functionality was introduced. Both samples were supplemented with a small catalytic amount of DMPA (∼1.0 wt% of total mixture) to accelerate the coupling reaction. The two distinct binary mixtures were transferred to cylindrical glass beakers and exposed to UV-irradiation delivering a constant light irradiance of 25 mW cm−2 (λmax = 365 nm) for a period of 6 hours which corresponds to an overall accumulated dose of 540 J cm−2. Variation in the incident UV-light intensity was observed not to exceed the 6.0% in the radial direction measured at the liquid surface. The photo-induced reactions were conducted at room temperature in the presence of ambient air under magnetic stirring (∼800 rpm). Diluting solvent was removed via rotor-evaporation at 50 °C followed by normal vacuum until residual ethyl acetate was eliminated. Resultant crude synthesis products were obtained as clear, slightly yellow, viscous liquids exhibiting a fresh citreous odor. All mixtures were prepared immediately prior to each experiment in order to ensure reproducibility of sample history.
2.3.2. Determination of conversion. 1H NMR and FT-Raman spectroscopies were used for the evaluation of thiol–ene conversions. Integral areas for the unsaturation proton signals observed at 4.7 ppm (exocyclic, vinylidene) and 5.4 ppm (endocyclic, trisubstituted) were normalized against the protons of (–CH2–O–) in the alkoxyl group from each thiol compound located at 4.1 ppm (signal c of Fig. 5); whereas in FT-Raman spectra, the thiol band (2610–2530 cm−1), exo-olefinic band (1645 cm−1); and, endo-olefinic band (1678 cm−1) were normalized against the band characteristic of the ester carbonyl group centered at 1735 cm−1. Percent conversions of each chemical group into β-thioether (C–S) bonds were determined from the formula:| |
 | (1) |
where x denotes either normalized double-bond 1H integrations or FT-Raman band heights before (x0) and after reaction (xf). Discontinuous kinetic conversion data were subsequently changed into chemical species (or functional group) concentrations as reported previously.15
2.4. Simulation software and modeling
Numerical kinetic simulations of the detailed reaction mechanism delineated in Scheme 1 were performed using the general software application package – COPASI (COmplex PAthway SImulator) version 4.8 (build 35). This stand-alone program was designed for the kinetic modeling, simulation and analysis of biochemical network systems.19 To construct the computed output profiles describing the concentration of the chemical species, all elementary step reactions, initial reactant concentrations (which determine the initial stoichiometry of the reaction system) and individual rate coefficients were first entered and the program allowed solving the system of differential equations as a function of time. The continuity equations describing the new reaction mechanism were simultaneously integrated in COPASI using the deterministic algorithm LSODA (Livermore Solver for Ordinary Differential Equations) up to a final running time of 180 min. LSODA is a very robust adaptive step-size solver that calculates the stiffness of equations and dynamically switches the method of integration according to this measure.20 Simulation with COPASI brings along the advantage of bypassing the steady-state approximation often required to calculate the concentrations of intermediary radical species.
3. Results and discussion
3.1. Thiyl radical generation, termination and main assumptions
Formation of initiating thiyl radicals was induced either photochemically via photolysis of DMPA upon exposure to polychromatic UV-light (285 ≤ λ ≤ 370 nm), or from thermal decomposition of the azo-compound AIBN at 70 °C (t1/2 = 4.8 hours).21 Each method promotes the intramolecular cleavage of the starting initiator boosting the production of thiyl radicals (RS˙) that initiate the cycles of thiol–ene coupling reactions. The initiation process involves, in a first step, formation of nucleophilic carbon-centered radical fragments as primary radicals through monomolecular decomposition of an initiator followed by hydrogen-abstraction from a thiol group via a hydrogen-transfer reaction (elementary reaction steps (1) and (2) of Scheme 1). The reaction then proceeds through a series of propagation–chain-transfer and termination steps which characterize the conventional thiol–ene mechanism. The primary radical fragments also have the possibility of adding to the double-bonds of the alkene monomer, react with dissolved oxygen (quenching) or undergo self-reaction leading to formation of transient photon-absorbing molecules. Additionally, it is expected that during initiation the decomposition of photoinitiator will dominate over the photolysis of the RS–H bond and disulfides as these chemical moieties absorb primarily at ∼254 nm light22–25 and at this wavelength the relative UV-light intensity delivered by the lamp source lies below 10%. Although these secondary reactions are all expected to occur, their influence will not be accounted in the present mechanistic model since they can be considered of minor importance to the overall kinetics which is governed by the central body of propagation–chain-transfer reactions; i.e., Ri + Rt ≪ Rp + RCT. The quantum yield of photoinitiation, i.e., initiator efficiency (0 < φ(λ) < 1), is usually a time-dependent parameter in bulk reactions and polymerizations due to changes in medium viscosity with increasing conversion which also affects the value of the first-order decomposition rate constant, kd.26 Therefore, a constant efficiency factor of 0.5 is assumed which also rectifies for the occurrence of the above mentioned side-reactions of the ensuing benzoyl, α,α-dimethoxy-benzyl and methyl radicals not leading to hydrogen-transfer reactions.26 This means that only one active radical fragment, I˙, is produced on average upon photo-decomposition of DMPA. To reduce system complexity, we also consider that the primary and secondary carbon-radical centers (I˙) possess equal reactivity in the hydrogen-abstraction reaction step. Moreover, we avoided introducing in the model the homo- and hetero-coupling terminations involving the two possible intermediate carbon-radicals and their reaction with thiyl radicals because there are no absolute values of the intrinsic rate constants currently available in the literature. Finally, we adopt a value for the first-order decomposition rate constant, kd = 10−4 s−1, estimated in a previous reference.15
3.2. Monothiol model studies
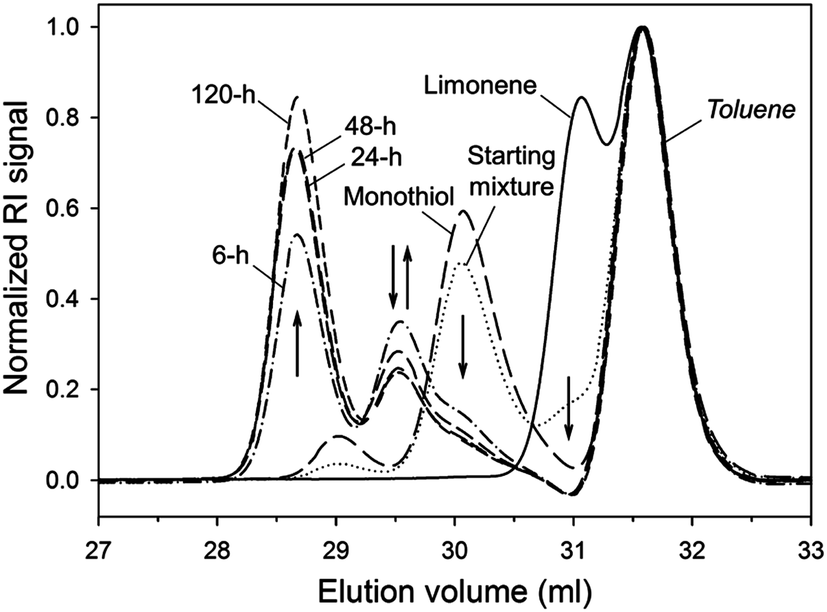
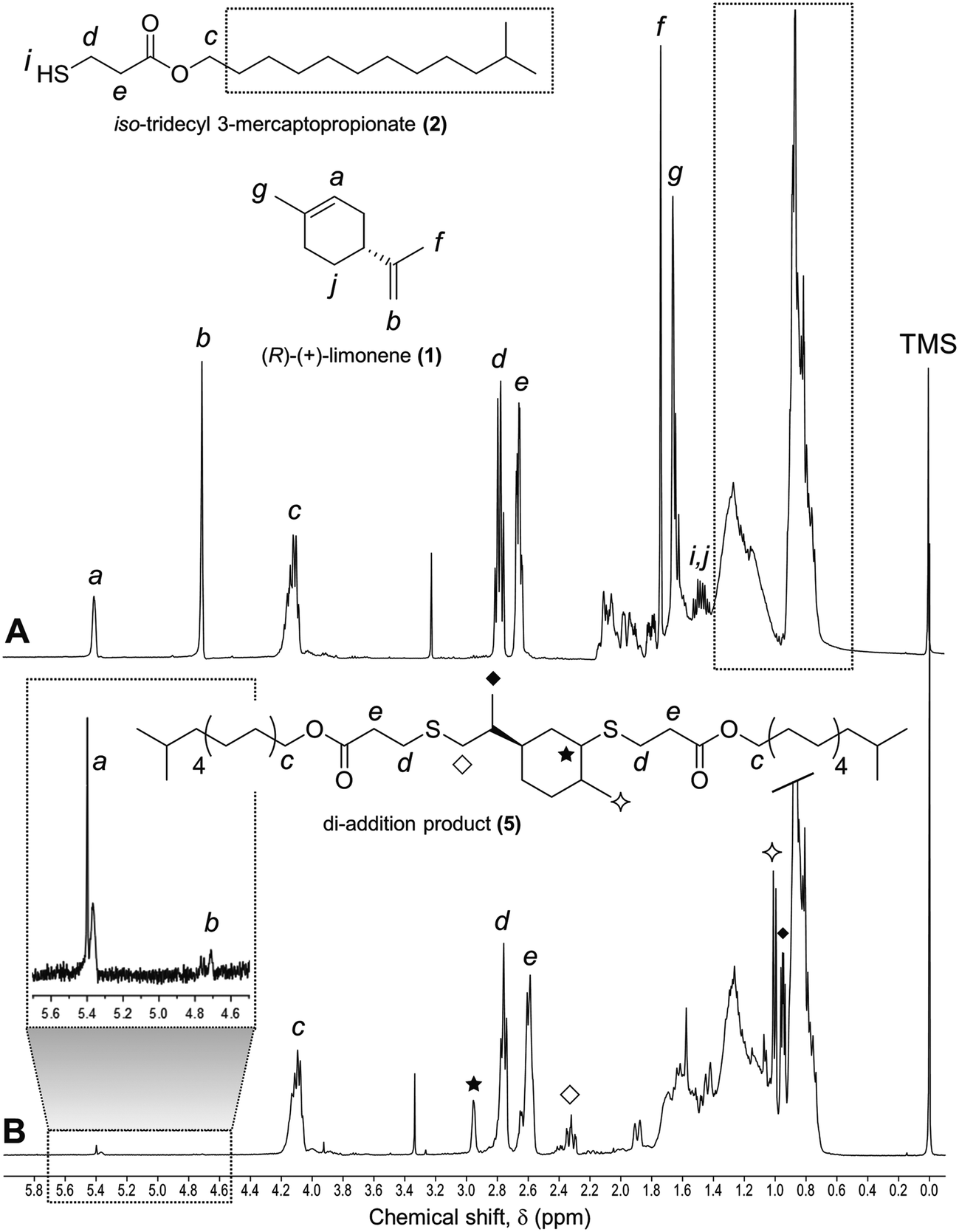
In preliminary experiments we treated (R)-(+)-limonene 1 with iso-tridecyl 3-mercaptopropionate 2 on a 1:1 mole ratio with respect to thiol and alkene functional groups under oxygen- and solvent-free conditions at ∼70 °C (Scheme 2). This monothiol reactant was chosen as prime model compound given its representative propionate ester moiety (RCH2OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OCH2CH2SH) characteristic of multifunctional thiols commonly used in thiol–ene polymerizations with the additional benefit of full miscibility with limonene at room temperature. The liquid bulk mixtures were supplemented with a small amount of the azo compound 2,2′-azo bis(2-methylpropionitrile) (AIBN) to speed up the production of initiating thiyl radicals. In order to protect limonene from prolonged exposure to oxidation and the deleterious radical trapping effect of oxygen commonly associated to free-radical reactions, the thermal reactions were carried out as closed systems in inert atmosphere after proper deoxygenation under vacuum. Analysis of the reaction mixtures over time by 1H NMR and FT-Raman spectroscopies (Fig. 1 and 2, respectively) revealed preferential thiol–ene coupling at the exo-vinylidene bond followed by a progressive increase in consumption of the endocyclic unsaturation. Consumption of the two distinct alkenes was accompanied by depletion of thiol functional groups leading to formation of the respective mono- and di-addition C–S products. The sequence of thiol–ene addition was monitored in parallel via DMF–SEC measurements as qualitatively displayed in Fig. 3. At long reaction times the formation of di-addition product (5, MW = 713.21 g mol−1) at 28.3–28.8 ml is favored at the expense of the two mono-additions (3 and 4) located at 29.3–29.8 ml which are predominantly formed in the initial period of the reaction. Since the two mono-addition products share exactly the same molecular weight (MW = 424.72 g mol−1), they could not be easily discriminated by SEC. Nonetheless, after a reaction period of 6 hours, practically all exo-vinylidene bonds were involved in thiol–ene coupling followed by slow gradual increase in conversion of the remaining trisubstituted double-bond (Fig. 1). Overall, and despite the observed differences in reactivity, a total final double-bond conversion of 90% was attained under these experimental conditions further illustrating the effectiveness of these two unsaturations in free-radical thiol–ene additions.
OCH2CH2SH) characteristic of multifunctional thiols commonly used in thiol–ene polymerizations with the additional benefit of full miscibility with limonene at room temperature. The liquid bulk mixtures were supplemented with a small amount of the azo compound 2,2′-azo bis(2-methylpropionitrile) (AIBN) to speed up the production of initiating thiyl radicals. In order to protect limonene from prolonged exposure to oxidation and the deleterious radical trapping effect of oxygen commonly associated to free-radical reactions, the thermal reactions were carried out as closed systems in inert atmosphere after proper deoxygenation under vacuum. Analysis of the reaction mixtures over time by 1H NMR and FT-Raman spectroscopies (Fig. 1 and 2, respectively) revealed preferential thiol–ene coupling at the exo-vinylidene bond followed by a progressive increase in consumption of the endocyclic unsaturation. Consumption of the two distinct alkenes was accompanied by depletion of thiol functional groups leading to formation of the respective mono- and di-addition C–S products. The sequence of thiol–ene addition was monitored in parallel via DMF–SEC measurements as qualitatively displayed in Fig. 3. At long reaction times the formation of di-addition product (5, MW = 713.21 g mol−1) at 28.3–28.8 ml is favored at the expense of the two mono-additions (3 and 4) located at 29.3–29.8 ml which are predominantly formed in the initial period of the reaction. Since the two mono-addition products share exactly the same molecular weight (MW = 424.72 g mol−1), they could not be easily discriminated by SEC. Nonetheless, after a reaction period of 6 hours, practically all exo-vinylidene bonds were involved in thiol–ene coupling followed by slow gradual increase in conversion of the remaining trisubstituted double-bond (Fig. 1). Overall, and despite the observed differences in reactivity, a total final double-bond conversion of 90% was attained under these experimental conditions further illustrating the effectiveness of these two unsaturations in free-radical thiol–ene additions.
 |
| | Scheme 2 Typical mono-/di-addition products resulting from free-radical thiol–ene coupling between (R)-(+)-limonene (1) and iso-tridecyl 3-mercaptopropionate (2) as monothiol model compound. | |
 |
| | Fig. 1 Extent of functional group conversion for a reaction mixture comprised of limonene (1) and iso-tridecyl 3-mercaptopropionate (2) reacted on a 1:1 mole stoichiometry with respect to thiol–ene functional groups, as provided by 1H NMR (double-bonds) and FT-Raman (thiol). The reactions were conducted thermally with 2.0 wt% of AIBN in bulk at ∼70 °C in the absence of oxygen. Error bars represent one standard deviation about the mean value measured from three independent reactions. | |
 |
| | Fig. 2 FT-Raman spectra collection showing the time-evolution of thiol and ene functional groups from a starting 1:1 stoichiometric mixture composed of (R)-(+)-limonene (1) and iso-tridecyl 3-mercaptopropionate (2). (a) Overall spectra in comparison to the individual pure compounds; (b) enhancement of region I showing the depletion of thiol (2573 cm−1); and, (c) zoom-in of region II showing the disappearance of unsaturations with favored coupling at the exocyclic double-bond position (1645 cm−1). The carbonyl moiety (CO, 1735 cm−1) was used as internal reference band in the spectral normalization process (only for the thiol–ene spectra). | |
 |
| | Fig. 3 DMF–SEC elugram showing the development of mono- and di-addition products resulting from selective thiol–ene coupling of 1 with 2 under thermal reaction conditions. The small bands centered at 29 ml represent trace amounts of disulfide product. The flow-rate marker (toluene) was set at 31.5 ml. Arrows denote directions in consumption and/or formation. | |
To better evaluate the discriminative power of both double-bonds regarding thiol–ene additions we then performed the same model reaction this time by changing the co-monomers feed ratio from 2:1 to 1:1 (thiol–terpene molecular ratio). The reaction was conducted at room temperature with the co-reactants mixed in bulk under multi-wavelength photoinduced conditions (4.2 mW cm−2) with the photoinitiator DMPA (2.0 wt% of total mixture) in the presence of atmospheric air. The comparative results shown in Fig. 4 fully corroborate the preliminary kinetic data obtained from thermal reaction once again showing preferential thiyl-radical attack at the exo-olefinic bond followed by subsequent consumption of the endocyclic ene. Near quantitative yields of the final di-addition product (5) were attained after an extended period of 8 hours reaction from a starting equimolar stoichiometry in thiol–ene functional groups (Fig. 5). The end-product was obtained as a viscous colorless liquid.
 |
| | Fig. 4 Kinetic plots of experimental data (symbols) vs. simulated model predictions (lines) as a function of initial thiol–ene stoichiometry obtained from photoinduced reactions of the monofunctional system. (a) Stoichiometry of 1:0.5, (b) and (c) stoichiometry of 1:1. Stoichiometric mole ratios are reported with respect to thiol and ene functionalities. Legend: thiol (RSH; ○, dotted line), limonene (Lim; long-dashed line), endocyclic unsaturation (endo; □, dashed-dotted line); and, exocyclic unsaturation (exo; ◇, medium-dashed line). Mono- (P) and di-addition (D) products are depicted by the continuous solid lines. Temporal evolution of overall alkene functional groups (i.e., enes present both in limonene and di-addition products) were calculated as: [exo]t = [Lim]t + [P2]t and [endo]t = [Lim]t + [P1]t from computed limonene and individual mono-additions. | |
 |
| | Fig. 5 1H NMR (400 MHz, CDCl3) spectra and principal signal assignments of thiol–ene samples comprised of compounds 1 and 2 mixed at 1:1 functional group stoichiometry. (A) Before photoinduced reaction (control); (B) after a reaction period of 8 hours conducted at room temperature. Integration value of protons of signal (c) was used as reference peak for the calculation of double-bond conversion. | |
3.3. Synthesis and characterization of thiol–ene addition products
Photoinduced conversion of thiol–ene reactants into di-addition product 5 was greater than 90%, as determined by 1H NMR after 8 hours of reaction (Fig. 5). The lack of the doublet signal (b) at 4.5 ppm assigned to the terminal protons of the exo-vinylidene bond and significant reduction of signal (a) at 5.4 ppm attributed to the single proton of the 1,1,2-trisubstituted 1-methyl-cyclohexene group confirmed that thiol–ene coupling of 1 with 2 was successful. Direct disappearance of these two signals was accompanied by the loss of triplet signals (f) at 1.75 ppm and (g) at 1.65 ppm, characteristic of the methyl protons contiguous to each double-bond, respectively. Furthermore, proton signals (e) and (d) centered at 2.66 and 2.78 ppm correspondingly, adjacent to the thiol group (∼1.5 ppm), were slightly shifted followed by formation of two new neighboring signals: a singlet (solid star) at ∼3.0 ppm and a quadruplet (white diamond) at 2.25–2.5 ppm assigned to the CH proton at position 2 and CH2 at position 10, respectively. Two new peaks located at 0.98–1.20 ppm (white star) and 0.88–0.98 ppm (two doublets, solid diamond) resultant from the coupling reaction were also visible corresponding to the terminal methyl groups at structural positions 7 and 9, respectively. The numbering terminology of each nucleus used for the assignment of 1H NMR signals was adopted from a previous reference.12 The same pattern was observed for the thermal synthesis of 5 under equimolar functional group stoichiometry after a reaction period of 120 hours (spectral 1H NMR results not shown). Characterization by FT-Raman spectroscopy (Fig. 6) shows the total consumption of thiol and alkene (exo- and endo-) functional groups, which demonstrates the completeness of the thiol–ene coupling reaction involving these two reactants.
 |
| | Fig. 6 Normalized FT-Raman spectra of samples obtained from photoinduced thiol–ene reaction of monofunctional system mixed on a 1:1 thiol–ene functional group stoichiometry. (a) Before reaction (starting mixture at time zero); (b) after a reaction period of 8 hours conducted at room temperature. The carbonyl ester group (CO, 1735 cm−1) was used as internal reference band in the spectral normalization process. | |
3.4. Four-route linear cyclic mechanism
We have developed an extended mechanism for the free-radical thiol–ene reaction of D-limonene accounting for all four cycles of propagation–chain-transfer reactions (Scheme 1a/c). The new model is based on the fundamental two-route linear step-growth mechanism detailed in a previous paper15 which describes just the two primary thiol–ene coupling reactions at each double-bond of limonene (exo or endo) (Scheme 1b). The governing chemical equations for the new model are shown in Scheme 1a. The upper part outlines a series of eight propagation–chain-transfer steps involving reactants, products and intermediate species; whereas the bottom denotes a set of four overall coupling reactions defined by the system of elementary steps. Simultaneous consumption of ene functionalities via a competing chain-growth route is not considered here as the two exotic double-bonds occurring in limonene are not readily homopolymerizable because of their high electron-density character,21,27,28 although it clearly occurs in certain other types of thiol–ene polymerizations such as thiol–(meth)acrylate systems.11 For simplification purposes we also assume that all kinetic parameters remain constant during the course of the reaction. Even though the reaction was performed in bulk liquid conditions and the medium viscosity increases with conversion, the efficient homogenization imposed by agitation significantly reduces molecular diffusion without major impact on the rate parameters. Additionally, this reaction system does not allow formation of a tridimensional cross-linked network or production of extremely high molecular weight products due to the mono- and di-functional nature of the co-reactants, which should keep the rate parameters unaltered. Finally, all reactions are assumed to occur under isothermal (room temperature) conditions although it was verified a slight temperature increase in the bulk phase relative to the ambient temperature as result of the exothermicity of the coupling reaction5 together with the energy delivered during UV-irradiation. Other simplifying assumptions are detailed in an earlier reference.15
3.5. Mechanistic model simulations of the global kinetics
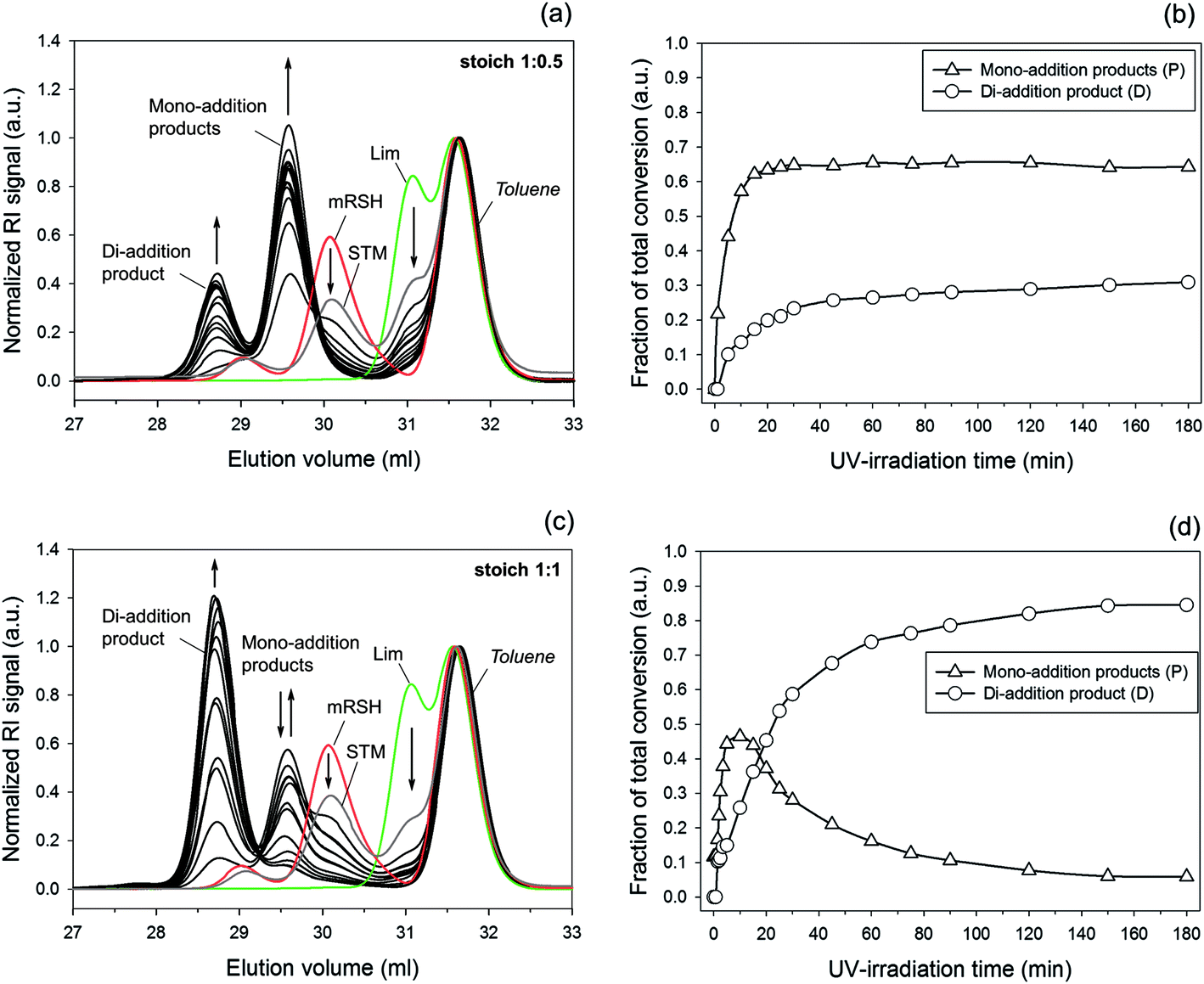
In our previous paper15 we observed the thiol–ene reaction with D-limonene to follow a second-order kinetics when the two co-monomers were charged in a 1:0.5 stoichiometry with respect to chemical groups in organic solution conditions; i.e., −rA = k*[A]2, with k* = kobs. This simple empirical rate law was found valid for the consumption of limonene as species and RSH functional groups as well as for the disappearance of individual alkenes (exo and endo); although in the latter case the overall rate parameter, kobs, was affected by a unique reactivity factor intrinsic to each unsaturated structure; i.e., k* = m1,2kobs. This led to a measured double-bond selectivity of 6.54 in favor of the exocyclic vinylidene bond. The value observed for the regioselectivity was further confirmed from numerical computations of the intrinsic rate coefficients applied to a simplified steady-state equation derived from the sequential four-step linear mechanism (Scheme 1b). To simplify the kinetic analysis, occurrence of secondary coupling reactions was neglected (cycles III and IV of Scheme 1c). As a result, Scheme 1b may be regarded as a ‘reduced’ case of Scheme 1a/c when the double-bond reactivity in secondary thiol–ene coupling is significantly reduced after primary coupling reactions; i.e., k3,4 ≈ 0. Yet, the two multi-substituted enes are located apart in the chemical structure of limonene, suggestive of k1,2 ≥ k3,4. When the reaction was performed in bulk liquid conditions, under an initial mole ratio of 1:0.5 (Fig. 7a/b) one clearly witnesses a small development of di-addition product (5) with reaction time, indicating an effective activation of cycles III and IV. By changing the feed ratio to equimolarity between thiol–ene functional groups, one observes significant effects on the overall reaction kinetics when compared with the previous stoichiometry as presented in Fig. 4 and 7, supported by experimental results given in Fig. S1 and S2 of the ESI.†
 |
| | Fig. 7 Normalized DMF–SEC elugrams (left side) and corresponding kinetic profiles (right side) showing the time-course evolution of mono- (3 + 4 = P) and di-addition (5 = D) products resulting from selective thiol–ene coupling under photochemical reaction conditions using two different stoichiometries with respect to reactive functional groups: (a) and (b) 1:0.5 mole ratio (1:1 thiol–terpene molecular ratio); (c) and (d) 1:1 mole ratio (2:1 thiol–terpene molecular ratio). Starting thiol–ene mixture (STM, grey line); Limonene (Lim 1, green line); and, Monothiol (mRSH 2, red line). The small bands centered at 29 ml represent trace amounts of disulfide product. The flow-rate marker (toluene) was set at 31.5 ml. Arrows denote directions in increase or decrease of the given coupled product(s). | |
Experimental reaction data obtained via 1H NMR/FT-Raman and DMF–SEC techniques were simulated numerically in COPASI using the rate coefficients optimized in a previous kinetic study.15 A value of kt = 3.0 × 108 M−1 s−1 was introduced for the thiyl-radical self-termination reaction, since the liquid medium viscosity increases by changing from solution to bulk conditions. Variation of the experimental conditions was required in order to follow the reaction via size-exclusion chromatography. Model simulations of both reactions confirm the validity of the new mechanism as qualitatively described by the excellent trend-shape agreement with the experimental profiles, despite the observed deviations (Fig. 4), which is also well correlated with the evolution of mono-/di-addition products displayed in Fig. 7. A small quantity of di-addition product (5) is always obtained for the 1:0.5 stoichiometry which is inevitably translated by a deviation between RSH functional groups consumed and reacted limonene as a species (Fig. 4a). This deviation was not apparent in our previous empirical kinetic study. For the reaction system corresponding to a stoichiometry of 1:1 we verify a distinct evolution pattern of the measured variables when compared to the previous case. The simulated reaction time corresponding to the maximum point of total mono-additions, tmax (Fig. 4b) appears to be in good agreement with the experimental time (Fig. 7) deducing that an order of magnitude of 108 for the termination rate constant, kt, seems appropriate in describing thiol–ene reaction systems operating in bulk conditions. Increasing kt by a factor of 10, as typically encountered in thiol–ene reactions carried out in organic solution,15,29,30 resulted in much lower reaction rates (results not shown). Three principal kinetic regions were identified with respect to the sequence pattern of coupled products formation based on the new mechanism: (i) before the maximum point is reached, at early instants in the reaction, only a residual amount of 5 is obtained (i.e., [D] ≈ 0) and the dominant coupling routes are described mostly by the propagation–chain-transfer cycles I and II leading to production of compounds 3 and 4, respectively; (ii) at the vicinity of the maximum point we are essentially in the presence of a kinetic transition from coupling cycles I and II to cycles III and IV, respectively; and, (iii) after the maximum point, the dominant pathways leading to formation of 5 are active (cycles III and IV) given that the large majority of limonene molecules have been formerly exhausted.
3.6. Kinetic effect of reaction stoichiometry
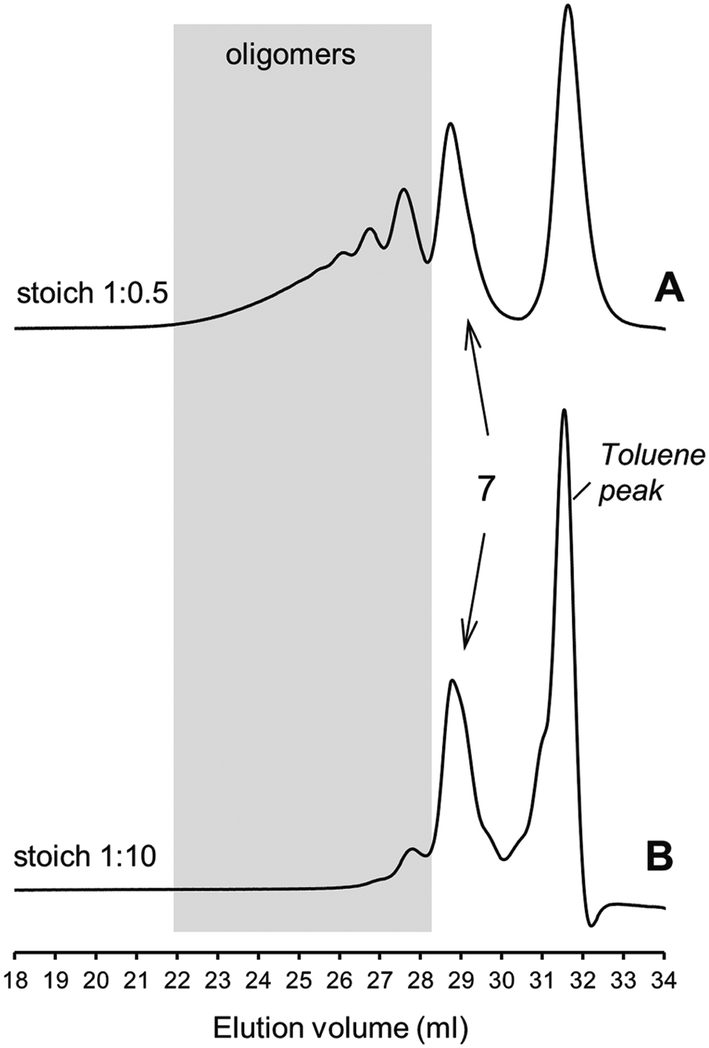
Once the validity of the four-route linear mechanism was confirmed we proceeded to determine numerically the kinetic effect of a wider range of starting stoichiometries on the evolution of the observable variables in order to assess which initial reaction conditions are more favorable towards the synthesis of one-to-one coupled products selectively at each unsaturation. The kinetic analysis was performed by assuming that primary coupling at each double-bond does not influence the reactivity of the remaining unsaturations in subsequent coupling reactions leading to formation of 5. Again, the kinetic simulations were performed using the rate coefficients estimated in our previous study15 with kt = 3.0 × 108 M−1 s−1 and then varying systematically the input concentrations of thiol and ene functional groups adjusted for each given stoichiometry (initial conditions). For all cases, a starting molar concentration of photoinitiator of 0.07 mol L−1 (∼2.0 wt% of total reaction mixture) was chosen in order to assure that the reaction system would not run out of propagating thiyl radicals consumed via bimolecular self-termination reaction. The simulation results are compiled in Fig. 8. This figure offers a comprehensive overview about the effect of initial thiol–ene stoichiometry on the overall behavior of the reaction system omitting influence of temporal effects and, therefore, can be viewed as a rational guidance tool aimed at the design of thiol–ene systems based on limonene. Plots (a) and (b) demonstrate the excellent ability of numerical simulations in describing the kinetic data obtained from experiment, indicating that this modeling approach represents an important methodology to assist empirical determinations. Plots (c) and (d) are complementary and represent final values of the variables after a running period of 180 min. As anticipated, by increasing the relative concentrations in D-limonene favors thiol–ene coupling at the exo-vinylidene bond along with growing suppression of 5; whereas, a relative increase in concentration of RSH groups foments production of 5 at the expense of monoaddition products (3 + 4). Towards very low thiol–ene stoichiometries, conversion of limonene becomes remarkably low because the terpene is present in relative excess while consumption of thiol achieves the highest value since it is the limiting reagent. Another prominent aspect related with this figure is that any set of experimental data points collected at a given stoichiometry can be interpreted just as a prolongation of experimental data acquired at lower stoichiometries. This feature is well expressed in plots (a) and (b) as the solid lines deviate just slightly from their dotted counterparts. Therefore, the chart represented in plot (c) is applicable to any workable stoichiometry without any substantial loss in accuracy. An average initial relative reactivity of (![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) exo/endo)0 = 9.74 ± 0.28, computed from the first-derivative of the individual alkene consumption profiles, was obtained from all stoichiometric pairs of exo-/endo-unsaturations. This value is consistent with previous determinations of the selectivity based on the reaction mechanism indicating that the relative double-bond reactivity is an intrinsic property of the reacting chemical groups and independent of any imposed stoichiometry. Another practical implication of the reaction mechanism straightly connected to differences in double-bond reactivity is that preferential thiol–ene coupling at the endocyclic double-bond can never be achieved or controlled just by means of adjusting the initial composition of co-reactants. An initial stoichiometric imbalance has, therefore, strong repercussions on the overall output kinetics, in special if an increment in limonene is targeted. This situation may very well turn advantageous synthetically toward the design of well-defined multifunctional ene-terminated macromonomers based on limonene and polyfunctional alkyl ester 3-mercaptopropionates without major interference of side-reactions such as thiol–ene coupling at the endo-olefinic bond and/or formation of large ‘hyperbranched-like’ oligomers derived from secondary coupling reactions (Scheme 3). Regarding multifunctional thiol–ene systems, the simulated results also predict that development of very high molecular weight oligomers is more prone to occur under stoichiometries of [RSH]0/[CC]0 ≥ 1; whereas at a [RSH]0/[CC]0 = 0.5 this effect is less pronounced. In order to confirm these expectations unambiguously, we have reacted isothermally the polyfunctional thiol TMPMP 6 with D-limonene 1 in ethyl acetate solution for an extended period of 6 hours and then analyzed the photo-reaction products by DMF–SEC after proper evaporation of the solvent. The elugrams plotted in Fig. 9 verify beyond any reasonable doubt the numerical predictions from the mechanism as demonstrated by the two very distinct patterns in molecular weight distribution of thioether coupled products. In elugram A is evident the development of a wider distribution in high molecular weight products as consequence of secondary coupling reactions, although macromonomer 7 is the main component assigned by the highest peak. In elugram B we observe a much narrower distribution in favor of mono-addition product 7. This pattern is dictated by the combinatory effect of double-bond selectivity and relative excess of limonene. The two residual peaks occurring at high molecular weights in elugram B (low elution volumes) demonstrate that despite the tentative manipulations in the starting co-reactant stoichiometry is not possible to obtain compound 7 in its pure form without minor intervention of secondary coupling reactions. Altogether, these results verify the adjustable but not controllable nature of free-radical thiol–ene additions involving multi-substituted alkenes in combination with the ‘non-click’ thiol–ene chemistry character associated to such olefins.
exo/endo)0 = 9.74 ± 0.28, computed from the first-derivative of the individual alkene consumption profiles, was obtained from all stoichiometric pairs of exo-/endo-unsaturations. This value is consistent with previous determinations of the selectivity based on the reaction mechanism indicating that the relative double-bond reactivity is an intrinsic property of the reacting chemical groups and independent of any imposed stoichiometry. Another practical implication of the reaction mechanism straightly connected to differences in double-bond reactivity is that preferential thiol–ene coupling at the endocyclic double-bond can never be achieved or controlled just by means of adjusting the initial composition of co-reactants. An initial stoichiometric imbalance has, therefore, strong repercussions on the overall output kinetics, in special if an increment in limonene is targeted. This situation may very well turn advantageous synthetically toward the design of well-defined multifunctional ene-terminated macromonomers based on limonene and polyfunctional alkyl ester 3-mercaptopropionates without major interference of side-reactions such as thiol–ene coupling at the endo-olefinic bond and/or formation of large ‘hyperbranched-like’ oligomers derived from secondary coupling reactions (Scheme 3). Regarding multifunctional thiol–ene systems, the simulated results also predict that development of very high molecular weight oligomers is more prone to occur under stoichiometries of [RSH]0/[CC]0 ≥ 1; whereas at a [RSH]0/[CC]0 = 0.5 this effect is less pronounced. In order to confirm these expectations unambiguously, we have reacted isothermally the polyfunctional thiol TMPMP 6 with D-limonene 1 in ethyl acetate solution for an extended period of 6 hours and then analyzed the photo-reaction products by DMF–SEC after proper evaporation of the solvent. The elugrams plotted in Fig. 9 verify beyond any reasonable doubt the numerical predictions from the mechanism as demonstrated by the two very distinct patterns in molecular weight distribution of thioether coupled products. In elugram A is evident the development of a wider distribution in high molecular weight products as consequence of secondary coupling reactions, although macromonomer 7 is the main component assigned by the highest peak. In elugram B we observe a much narrower distribution in favor of mono-addition product 7. This pattern is dictated by the combinatory effect of double-bond selectivity and relative excess of limonene. The two residual peaks occurring at high molecular weights in elugram B (low elution volumes) demonstrate that despite the tentative manipulations in the starting co-reactant stoichiometry is not possible to obtain compound 7 in its pure form without minor intervention of secondary coupling reactions. Altogether, these results verify the adjustable but not controllable nature of free-radical thiol–ene additions involving multi-substituted alkenes in combination with the ‘non-click’ thiol–ene chemistry character associated to such olefins.
 |
| | Fig. 8 Concentration–conversion plots a/b and evolution of conversion as a function of total ene conversion or thiol–ene stoichiometry c/d. Experimental (symbols) vs. simulated (lines) data for a composition mole ratio of (a) 1:2 (i.e., 1:0.5), and (b) 1:1 with respect to thiol–ene functionalities. (c) General simulation chart covering a stoichiometry range of 1:20 ≤ thiol:ene ≤ 10:1. Full-range plot-lines represent a maximal thiol–ene stoichiometry of 20:1. (d) Effect of stoichiometry on the final conversion of variables. Individual symbols as data points in plots (c) and (d) denote final computed concentrations or conversions (at t = 180 min) of each variable at a given initial thiol–ene stoichiometry. [DMPA]0 = 0.07 M (∼2.0 wt% of total amount of reactants) in all simulations. Symbols (+) and (−) in plot (d) denote positive or negative conversions of a given variable, respectively. | |
 |
| | Scheme 3 Selective thiol–ene coupling between (R)-(+)-limonene (1) and the trithiol TMP-trimercaptopropionate (6). Starting thiol–ene stoichiometry: (a) low thiol–ene stoichiometries (excess of limonene); (b) high thiol–ene stoichiometries (excess of thiol). Photoinitiator (DMPA, 2.0 wt%), solvent (EtOAc, 50 wt%), UV-light irradiance: 25 mW cm−2 (dose: 540 J cm−2), agitation speed: 800 rpm. | |
 |
| | Fig. 9 Qualitative DMF–SEC elugrams for the multifunctional system based on the photoinitiated reaction of (R)-(+)-limonene (1) with TMP-trimercaptopropionate (6) using two different thiol-to-ene stoichiometries. In elugram B the small band shoulders located at 29.5, 30.5 and 31.0 ml represent vestigial amounts of partially coupled products, unreacted trithiol and limonene, respectively. Thiol:ene = x:y (functional group stoichiometry). Shaded grey region denotes occurrence of high molecular weight ‘hyperbranched-like’ oligomers. | |
4. Conclusions
In an ongoing effort to integrate monomers from renewable resources into cross-linked thiol–ene networks we have continued investigating the kinetics of free-radical thiol–ene additions between D-limonene and two synthetic mono-/tri-functional thiol monomers containing a propionate ester moiety. Monothiol model reactions conducted either thermally or photochemically indicated preferential thiol–ene coupling at the exocyclic vinylidene bond of limonene over the endocyclic ene in bulk liquid conditions. An extended mechanistic scheme accounting both for primary and secondary coupling reactions was conceived for limonene and tested numerically in the application software COPASI reproducing with good fidelity the experimental kinetic data. The simulations covered a broad range of initial concentrations for testing the impact of thiol–ene stoichiometry on the output concentration profiles and outcome of the reaction. Manipulations in the stoichiometry show that coupling at the exo-vinylidene bond can be addressed regioselectively by increasing the relative concentration of monoterpene. The superior rates attained via UV-irradiation over thermally induced reactions, along with negligible occurrence of secondary reactions, makes this method suitable for green industrial synthesis processes using limonene and multifunctional thiols. The knowledge resulting from this study provides researchers with a fundamental kinetic platform for designing thiol–ene systems based on limonene and alkyl ester 3-mercaptopropionates.
Acknowledgements
We kindly acknowledge financial support from the Swedish Research Council (Vetenskapsrådet), grant # 621-2007-5723.
References
- M. N. Belgacem and A. Gandini, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008 Search PubMed.
- A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- N. Moszner, W. Schoeb and V. Rheinberger, Polym. Bull., 1996, 37, 289–295 CrossRef CAS.
- P. S. Kulkarni, C. Brazinha, C. A. M. Afonso and J. G. Crespo, Green Chem., 2010, 12, 1990–1994 RSC.
- S. E. Kasprzak, B. Martin, T. Raj and K. Gall, Polymer, 2009, 50, 5549–5558 CrossRef CAS PubMed.
- C. Nilsson, Ph. D. Thesis, KTH Royal Institute of Technology, 2008.
- L. Lecamp, F. Houllier, B. Youssef and C. Bunel, Polymer, 2001, 42, 2727–2736 CrossRef CAS.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- M. Stemmelen, F. Pessel, V. Lapinte, S. Caillol, J. P. Habas and J. J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2434–2444 CrossRef CAS.
- M. Black and J. W. Rawlins, Eur. Polym. J., 2009, 45, 1433–1441 CrossRef CAS PubMed.
- M. Claudino, M. Jonsson and M. Johansson, RSC Adv., 2013, 3, 11021–11034 RSC.
- M. Claudino, M. Johansson and M. Jonsson, Eur. Polym. J., 2010, 46, 2321–2332 CrossRef CAS PubMed.
- M. Claudino, I. van der Meulen, S. Trey, M. Jonsson, A. Heise and M. Johnasson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 16–24 CrossRef CAS.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Macromolecules, 2003, 36, 7964–7969 CrossRef CAS.
- S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
- L. A. H. Petzold, LSODA (Livermore Solver of Ordinary Differential Equations), Livermore, CA, 1997 Search PubMed.
- G. Odian, in Principles of Polymerization, ed. N. J. Hoboken, John Wiley & Sons, Inc., New Jersey, 4th edn, 2004, ch. 3, pp. 135–162 Search PubMed.
- N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3311–3319 CrossRef CAS.
- N. B. Cramer, S. K. Reddy, M. Cole, C. Hoyle and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5817–5826 CrossRef CAS.
- A. K. O'Brien, N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2007–2014 CrossRef CAS.
- N. A. Rosenthal and G. Oster, J. Am. Chem. Soc., 1961, 83, 4445–4448 CrossRef CAS.
- D. L. Kurdikar and N. A. Peppas, Macromolecules, 1994, 27, 733–738 CrossRef CAS.
- H. Morinaga, Y. Kiyokawa, M. Kataoka, J. Masuda and D. Nagai, Polym. Bull., 2013, 70, 1113–1123 CrossRef CAS.
- A. Singh and M. Kamal, J. Appl. Polym. Sci., 2012, 125, 1456–1459 CrossRef CAS.
- C. Chatgilialoglu, A. Samadi, M. Guerra and H. Fischer, ChemPhysChem, 2005, 6, 286–291 CrossRef CAS PubMed.
- C. Chatgilialoglu, A. Altieri and H. Fischer, J. Am. Chem. Soc., 2002, 124, 12816–12823 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47922f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a