
New layer-by-layer Nb2O5–TiO2 film as an effective underlayer in dye-sensitised solar cells
Abstract
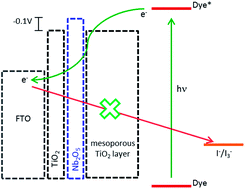
Highly efficient all-inorganic TiO2–Nb2O5 underlayers for dye-sensitised solar cell applications were produced by the layer-by-layer technique (LbL). TiO2 and Nb2O5 nanoparticles were prepared by the sol–gel method under acidic and alkaline conditions, respectively. The LbL films exhibited a very compact and homogeneous surface, as shown in FESEM and AFM images, which ensured a physical barrier between the electrolyte and the FTO surface, decreasing the dark current at this interface. Moreover, the rough film surface improved the physical interaction between the mesoporous TiO2 layer and the conductive substrate. The Ti(IV)/Nb(V) molar ratio in the films was 1.6, as determined by XPS, and it is controlled by the pH employed during the deposition process. The relative concentration of nanoparticles in the film plays a major role in its electronic properties: a higher TiO2 concentration allows an efficient transport of photoinjected electrons. Additionally, the presence of Nb2O5 nanoparticles imposes an electronic barrier for charge transfer from the FTO to the electrolyte, as shown by electrochemical impedance spectroscopy. Thus, all the DSC photoelectrochemical parameters increased, leading to an impressive improvement in the overall conversion efficiency.
 Please wait while we load your content...
Please wait while we load your content...

