Facile functionalization of FK506 for biological studies by the thiol–ene ‘click’ reaction†
Zhi-Fo Guo,
Roushu Zhang and
Fu-Sen Liang*
Department of Chemistry and Chemical Biology, University of New Mexico, Albuquerque, New Mexico 87131, USA. E-mail: fsliang@unm.edu; Fax: +1-505-277-2609; Tel: +1-505-277-1703
First published on 21st January 2014
Abstract
An efficient one-step functionalization of FK506 by the thiol–ene click (TEC) reaction is reported. This approach, which enables rapid and quantitative generation of bioactive FK1012 and FK506 derivatives, should facilitate biomedical applications of FK506-coupled molecules and expand the scope of the TEC reaction in natural product semi-synthesis.
FK506 (1 in Scheme 1) is a natural product that binds specifically to the abundant and ubiquitous mammalian protein, FK506-binding protein 12 (FKBP12).1 This unique recognition property has been advantageously used in various biomedical research applications. As an FDA approved immunosuppressant agent (Tacrolimus), its biological activity comes from the blocking of the calcineurin phosphatase active site by the composite surface of the FK506/FKBP12 complex.1 In addition, various applications have been reported that take advantage of ligand-mediated recruitment of FKBP12. In one, coupling two FK506 molecules with a linker creates a homo-dimerizer, FK1012, which has been used in chemically induced proximity (CIP) technology2 to promote dimerization of FKBP12-tagged proteins and control a variety of downstream cellular processes.3 Another application involves linking FK506 or the synthetic ligand of FKBP (SLF)4 to other protein binding ligands to generate hetero-dimerizers that induce the association of FKBP12-fused proteins with other proteins.5 Conjugating FKBP12-binding ligands to other small molecules leads to the formation of small molecule-FKBP12 complexes in which the small molecule components gain modified properties. For example, formation of these complexes adds additional bulkiness to the small molecule needed to block protein–protein interaction,6a increases the recognition surface that alters binding affinity and selectivity,6b or modulates the cellular context-dependent protein recognition.6c Generation of small molecule-FKBP12 complexes also protects small molecules from the cytochrome p450-mediated metabolic clearance and increases their half-lives in vivo.7 Owing to the diverse applications of ligand-induced FKBP12 recruitment, methods that enable robust and efficient conjugation of FKBP12-recruiting ligands to small molecules are highly desirable.
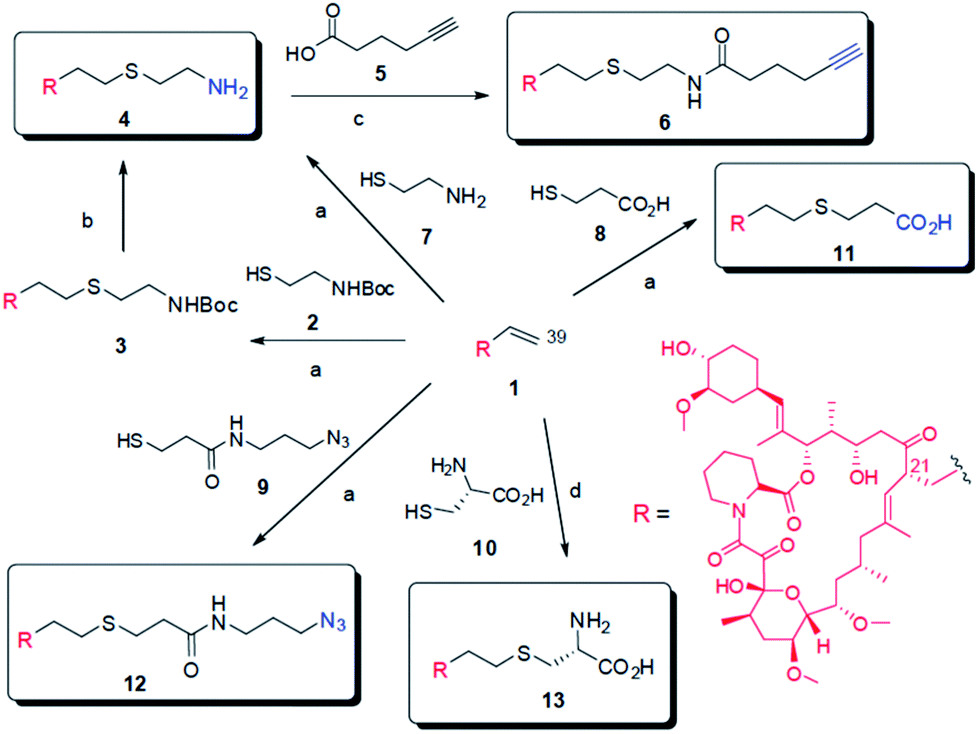 | ||
| Scheme 1 Synthesis of FK506 derivatives. The general TEC condition: l eq. FK506, 1 eq. thiols and 0.05 eq. of DPAP dissolved in indicated solvents under UV (λ365 nm) at room temperature for 15 minutes. aTEC in DCM; bTFA/DCM; cEDCI, HOBt, NEt3; dTEC in MeOH/H2O. | ||
Although SLF has a 10-fold decreased affinity for FKBP12 when compared to that of FK506,4 it is the most commonly used FKBP12-recruiting ligand to link to other molecules. The main reason for the extensive use of SLF is the ease of chemical derivatization through its aniline or carboxylate group.4b,6a,6c,8a,8b However, SLF is extremely expensive ($10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 360 per g, Cayman Chemical Company) and an 8- to 12 step route is needed for its synthesis in only moderate yields.4a,4b,8a On the contrary, FK506 is commercially available at a much lower cost ($160 per g, China Langchem Inc.) and has a much higher FKBP12-binding affinity. Nevertheless, its structural complexity and the lack of easily modifiable chemical handles make FK506 derivatives less accessible.
360 per g, Cayman Chemical Company) and an 8- to 12 step route is needed for its synthesis in only moderate yields.4a,4b,8a On the contrary, FK506 is commercially available at a much lower cost ($160 per g, China Langchem Inc.) and has a much higher FKBP12-binding affinity. Nevertheless, its structural complexity and the lack of easily modifiable chemical handles make FK506 derivatives less accessible.
Thus far, several methods have been reported to modify FK506 at the C39 position through chemical manipulation of the C21 allyl group. Modifications at this site, which is involved in calcineurin recognition,9 have been shown to destroy calcineurin binding without impacting the affinity for FKBP12.10 Some of the methods employed for FK506 modification require multi-step transformations, multiple protecting group manipulations, and proceed in low overall yields.3a Others, which employ Grubbs' type cross metathesis to convert the exocyclic alkene moiety of FK506 into other functional groups, take place in a single step with modest yields (34–49%).10,11a–c One recent approach using microwave-assisted cross metathesis generated FK506 derivatives in moderate to excellent yields (35 to over 95%) within minutes at high temperatures (ca. 150 °C).11d However, the requirement for special microwave instrumentation and the inconsistencies among different microwave apparatuses limit the general applicability of this method.
Thiol–ene click (TEC) reaction is a century-old reaction that occurs between thiols and alkenes to give anti-markovnikov thioethers through a well-established free-radical mechanistic pathway.12 Several applications of TEC reaction in polymer synthesis and carbohydrate/protein modifications have been reported.13 It tolerates various functional groups and proceeds rapidly under mild, aerobic conditions. Importantly, neither expensive or potentially toxic metal catalysts nor any special apparatus are required for this photo-initiated reaction. TEC reaction is considered as photo-click chemistry because this reaction has the same robustness, selectivity, simplicity and mildness as click chemistry.14
Owing to these features, the TEC reaction is a potentially attractive method to modify FK506 through its unique exocyclic alkene group. In the study described below, we show that several common chemical handles used in deriving bioactive molecules, including an amine, carboxylic acid, alkyne and azide, can be easily and rapidly installed onto FK506 by using TEC reactions. The processes are initiated by using a handheld UV lamp without the need of special photo-reactors and proceed smoothly in various organic solvents or with water as a co-solvent. Finally, we also demonstrated that the bioactive FK1012 can be synthesized in a single step in quantitative yield within minutes.
To investigate the feasibility of using the TEC reaction to derivatize FK506, we first examined the reaction of unprotected FK506 with the simple thiol, Boc-cysteamine 2 (Scheme 1). For this purpose, the mixture of equal molar ratio of FK506 and 2 along with a 0.05 equivalent (eq.) of the photo initiator 2,2-dimethoxy-2-phenylacetophenone (DPAP) was dissolved in dichloromethane (DCM) and irradiated using a handheld UV lamp (λmax 365 nm) (Fig. S1†). The reaction was carried out at room temperature under normal atmospheric conditions without any special degassing or drying procedure. Notably, the reaction proceeded rapidly with thin-layer chromatography (TLC) analysis showing that complete conversion occurred within 15 min. The product 3 was easily purified by flash chromatography in near quantitative yield. Analysis of the NMR and mass spectrometric data verified that compound 3 was generated through regioselective exocyclic alkene coupling.
To test the potential of using TEC for coupling chemical moieties that may have varied solvent solubility, TEC reactions between FK506 and 2 were carried out in DCM, toluene, or methanol for 15 min under the described conditions. All three reactions, taking place in solvents with very different polarities and protic natures, gave the coupling product 3 in near quantitative isolated yields (Table 1). To probe the rates, the above reactions were monitored using high-performance liquid chromatography (HPLC). In all cases, significant conversions were observed within 10 min and the reactions were complete within 15–20 min (Fig. S2†). The Boc-group of compound 3 can be removed to give the corresponding amine 4, which can be readily converted into other desired functional groups. Employing this approach, we prepared the FK506 alkyne derivative 6 through amide coupling with the alkyne-containing acid 5. The alkyne group in 6 can be used to introduce other fragments through copper-catalyzed click chemistry.13a
To enable more efficient chemical derivatization of FK506, we investigated the feasibility of directly coupling unprotected FK506 with thiols like cysteamine 7, 3-thiopropanoic acid 8, azido-thiol 9 and cysteine 10, which contain respective unprotected amine, carboxylic acid, azide and amino acid moieties. These TEC reactions were performed in solvents in which the reactants have good solubility (Scheme 1). In the case of the reaction with cysteine, water was added as co-solvent to obtain the desired solubility and to evaluate the effectiveness of TEC reactions in environmental friendly and biologically compatible conditions. In all cases, the reactions proceeded efficiently to give the desired adducts 4 and 11–13 in near quantitative yield within minutes (Table 1). These examples showcase the generality of TEC reactions in modifying FK506 and potentially other complex natural products containing terminal alkene groups.
To demonstrate the usefulness of TEC reactions for FK506 modification, a new FK1012 was synthesized. The previously reported syntheses of FK1012 variants either involve multi-step transformations with 18% overall yields,3a or a single-step cross metathesis with 50% yield with a 22 h reaction time.11a The FK1012s generated in these ways contained different linkers to bridge two FK506 molecules and had comparable biological activities.3a,11a Using dithiothreitol (DTT) 14 as the linker, 2 eq. of FK506 and 1 eq. of DTT were coupled under the TEC reaction condition to give FK1012-DT 15 in one step within 15 min in quantitative yield (Scheme 2a). The reported FK1012-EZ 16, a FK1012 possessing an alkene linker, was also prepared (Scheme 2b)11a so that we could compare the abilities of 15 and 16 to induce protein dimerization in an inducible luciferase transcription assay (Fig. 1a). For the assay, two DNA constructs, each encoding an FKBP12-fusion protein, were prepared. One construct expresses the yeast GAL4 DNA binding domain (GAL4DBD) fused to 3 copies of FKBP12 and the other expresses the herpes simplex virus VP16 transactivation domain (VPAD) linked to 2 copies of FKBP12 (Fig. 1a). A reporter construct containing 5 copies of the upstream activation sequence (UAS) at the promoter region for the luciferase gene was also used.15 Once these DNA plasmids are introduced into cells, the expressed GAL4DBD-FKBP fusion protein will be targeted to the UAS but cannot activate luciferase expression unless FK1012 is present to recruit the expressed VPAD-FKBP protein to the luciferase gene. To test the activity of 15 and 16, the above plasmids were co-transfected into CHO cells 24 h before adding 200 nM FK506, 100 nM 15, 100 nM 16, or without drugs. The cells were incubated for 10 h before being harvested and subjected to the luciferase assay. As expected, a 6 to 10 fold induction (based on the no drug sample) of luciferase occurred only when 15 or 16 was added (Fig. 1b). Induction was not observed when the monomer FK506 was added. These results clearly demonstrate that the much more efficiently synthesized FK1012-DT 15 is biologically active at a level comparable or better than those of previously reported analogs.
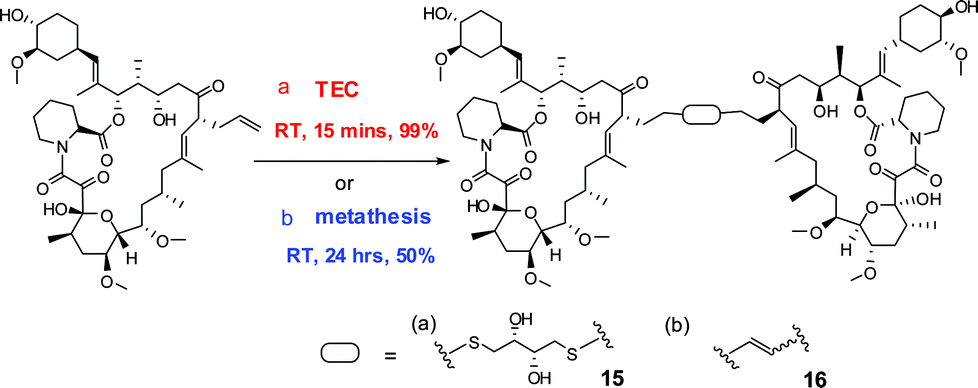 | ||
| Scheme 2 Synthesis of FK1012s. (a) 0.5 eq. DTT, cat. DPAP, DCM, UV365 nm; (b) 10% Grubbs (II) cat, DCM. | ||
 | ||
| Fig. 1 (a) FK1012-induced luciferase expression and DNA constructs for the asays; (b) induction fold change of luciferase. The results were generated from three experiments. | ||
In conclusion, we have developed a new, efficient thiol–ene click protocol for the facile functionalization of FK506. Under mild reaction conditions, commonly used chemical handles can be conveniently installed on FK506 without impacting FKBP12 binding and enable the production of complex bioactive molecules (e.g. FK1012). This method provides an attractive alternative to existing approaches for the derivatization of FK506. We envision that FK506 has the potential of serving as a general and useful installation for various biologically active small molecules or macromolecules that recruit endogenous FKBP12 to modulate the activity, binding selectivity or stability of conjugated molecules. This new and efficient FK506 modification method should significantly facilitate these applications. To our knowledge, this study is the first to apply the TEC reaction to natural product semi-synthesis. We expect that the scope of TEC reactions will expand beyond current applications to the semi-synthesis of structurally complex natural products.
Acknowledgements
We thank UNM for financial support and Dr Wei Wang for helpful discussions.Notes and references
- (a) J. Liu, J. D. Farmer, W. S. Lane, J. Friedman, J. Weissman and S. L. Schreiber, Cell, 1991, 66, 807 CrossRef CAS; (b) S. Ho, N. Clipstone, L. Timmermann, J. Northrop, I. A. Graef, D. Fiorentino, J. Nourse and G. R. Crabtree, Clin. Immunol. Immunopathol., 1996, 80, S40 CrossRef CAS.
- (a) J. E. Gestwicki and P. S. Marinec, Comb. Chem. High Throughput Screening, 2007, 10, 667 CrossRef CAS; (b) A. Fegan, B. White, J. C. T. Carlson and C. R. Wagner, Chem. Rev., 2010, 110, 3315 CrossRef CAS PubMed; (c) R. DeRose, T. Myyamoto and T. Inoue, Pflugers Arch., 2013, 465, 409 CrossRef CAS PubMed.
- (a) D. M. Spencer, T. J. Wandless, S. L. Schreiber and G. R. Crabtree, Science, 1993, 262, 1019 CAS; (b) L. J. Holsinger, D. M. Spencer, D. J. Austin, S. L. Schreiber and G. R. Crabtree, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9810 CrossRef CAS; (c) S. Ho, S. R. Biggar, D. M. Spencer, S. L. Schreiber and G. R. Crabtree, Nature, 1996, 382, 822 CrossRef CAS PubMed.
- (a) D. A. Holt, J. I. Luengo, D. S. Yamashita, H.-J. Oh, A. L. Konialian, H.-K. Yen, L. W. Rozamus, M. Brandt, M. J. Bossard, M. A. Levy, D. S. Eggelston, J. Liang, L. W. Schultz, T. J. Stout and J. Clardy, J. Am. Chem. Soc., 1993, 115, 9925 CrossRef CAS; (b) T. Clackson, W. Yang, L. W. Rozamus, M. Hatada, J. F. Amara, C. T. Rollins, L. F. Stevenson, S. R. Magari, S. A. Wood, N. L. Courage, X. Lu, F. Cerasoli Jr, M. Gilman and D. A. Holt, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10437 CrossRef CAS.
- J. L. Czlapinski, M. W. Schelle, L. W. Miller, S. T. Laughlin, J. J. Kohler, V. W. Cornish and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 13186 CrossRef CAS PubMed.
- (a) J. E. Gestwicki, G. R. Crabtree and I. A. Graef, Science, 2004, 306, 865 CrossRef CAS PubMed; (b) R. Briesewitz, G. T. Ray, T. J. Wandless and G. R. Crabtree, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1953 CrossRef CAS; (c) P. D. Braun, K. T. Barglow, Y.-M. Lin, T. Akompong, R. Briesewitz, G. T. Ray, K. Haldar and T. J. Wandless, J. Am. Chem. Soc., 2003, 125, 7575 CrossRef CAS PubMed.
- (a) P. S. Marinec, J. K. Lancia and J. E. Gestwicki, Mol. BioSyst., 2008, 4, 571 RSC; (b) P. S. Marinec, L. Chen, K. J. Barr, M. W. Mutz and J. E. Gestwicki, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1336 CrossRef CAS PubMed.
- (a) J. F. Amara, T. Clackson, V. M. Rivera, T. Guo, T. Keenan, S. Natesan, R. Pollock, W. Yang, N. L. Courage, D. A. Holt and M. Gilman, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10618 CrossRef CAS; (b) T. Keenan, D. R. Yaeger, N. L. Courage, C. T. Rollins, M. E. Pavone, V. M. Rivera, W. Yang, T. Guo, J. F. Amara, T. Clackson, M. Gilman and D. A. Holt, Bioorg. Med. Chem., 1998, 6, 1309 CrossRef CAS.
- J. P. Griffith, J. L. Kim, E. E. Kim, M. D. Sintchak, J. A. Thomson, M. J. Fitzgibbon, M. A. Fleming, P. R. Caron, K. Hsiao and M. A. Navia, Cell, 1995, 82, 507 CrossRef CAS.
- P. A. Clemons, B. G. Gladstone, A. Seth, E. D. Chao, M. A. Foley and S. L. Schreiber, Chem. Biol., 2002, 9, 49 CrossRef CAS.
- (a) S. T. Diver and S. L. Schreiber, J. Am. Chem. Soc., 1997, 119, 5106 CrossRef CAS; (b) A. M. Piggott, A. M. Kriegel, R. D. Willows and P. Karuso, Bioorg. Med. Chem., 2009, 17, 6841 CrossRef CAS PubMed; (c) K. Koide, J. M. Finkelstein, Z. Ball and G. L. Verdine, J. Am. Chem. Soc., 2001, 123, 398 CrossRef CAS PubMed; (d) P. S. Marinec, C. G. Evans, G. S. Gibbons, M. A. Tarnowski, D. L. Overbeek and J. E. Gestwicki, Bioorg. Med. Chem., 2009, 17, 5763 CrossRef CAS PubMed.
- (a) T. Posner, Chem. Ber., 1905, 38, 646 CrossRef; (b) M. S. Kharasch, A. T. Read and F. R. Mayo, Chem. Ind., 1938, 57, 752 CrossRef.
- (a) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed; (b) A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573 RSC.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 40, 2004 CrossRef CAS; (b) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666 CrossRef CAS PubMed; (c) A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995 CrossRef CAS PubMed; (d) R. K. V. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828 CrossRef CAS PubMed.
- F.-S. Liang, W. Q. Ho and G. R. Crabtree, Sci. Signaling, 2011, 4, rs2 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47867j |
| This journal is © The Royal Society of Chemistry 2014 |