
A non-platonic M4L4 complex constructed using heterotopic ligands†
Abstract
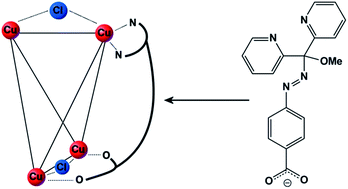
An unusual, chloride-capped M4L4 complex has been prepared using heterotopic ligands with carboxylate and dipyridyl coordinating sites. The incorporation of low symmetry, heterotopic ligands into the M4L4 architecture gives rise to a non-platonic complex with irregular triangular faces, demonstrating that the standard high-symmetry model traditionally adopted for synthesising such complexes is not a strict rule. The M4L4 compound is also observed by mass spectrometry in solution, yet shows no indication of host–guest behaviour in its small cavity. The assembly of the complex is assisted by a ‘belt’ of CH⋯π interactions.
 Please wait while we load your content...
Please wait while we load your content...

