 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Current-driven nitrate migration out of groundwater by using a bioelectrochemical system
Yiran Tonga and
Zhen He*b
aDepartment of Civil Engineering and Mechanics, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, USA
bVia Department of Civil and Environmental Engineering, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA. E-mail: zhenhe@vt.edu; Fax: +1 (540) 231-7916; Tel: +1 (540) 231-1346
First published on 3rd February 2014
Abstract
A bioelectrochemical system (BES) was designed to remove nitrate from groundwater with multiple barriers to prevent intrusion of undesired ions from its anolyte. Electric current was demonstrated to be a key force to transport nitrate ions, and OH− ions were identified as a major competitor with nitrate for anion migration. The balanced mass between the lost nitrate from groundwater and the accumulated nitrate in the concentrating chamber suggested that nitrate removal was mostly due to physical migration, rather than biological reduction. Despite several challenges, the results encourage the further investigation and development of this BES for in situ nitrate remediation from groundwater.
1 Introduction
Nitrate is one of the contaminants that appear in groundwater causing strong health and environmental concerns.1 Due to a lack of suitable electron donors and nitrate's mobility in groundwater, the removal of nitrate from groundwater in an effective and efficient way is a great challenge. The ex situ treatment methods such as pump-and-treat are energy intensive, while the in situ methods like chemical or biological nitrate reduction require addition of chemical compounds and stimulation of bacterial growth in groundwater that may cause further environmental problems.2 Thus, separating nitrate from groundwater with minimal influence on groundwater quality is of strong interest, and here we propose the use of a bioelectrochemical system (BES) to achieve this goal.As an emerging concept that can accomplish the removal of organic matters from water/wastewater while simultaneously producing electricity,3,4 BES has been studied for nitrate removal through bioelectrochemical nitrate reduction, mainly from wastewater.5 Several BES reactors were developed for groundwater nitrate remediation via extracting nitrate from groundwater for further biological reduction.6,7 A potential concern of using BES for in situ nitrate remediation, especially when low-grade substrates such as wastewater are used as an electron donor, is that the undesired ions in the anolyte could move into groundwater via ion exchange, thereby affecting groundwater quality. Although it was demonstrated that electricity generation may reduce the chance of this migration,7 the salt gradient between the anode and groundwater can still promote ion exchange and introduce ions that have a higher concentration in the anode solution into groundwater, which raises an alert that we need to prevent additional contamination while accomplishing nitrate removal. Having one layer of ion exchange membrane between groundwater and the anode of a BES (where microorganisms and complex solution are) can hardly prevent ion transport between the two.
In this study, we have designed a new BES with multiple barriers between the anolyte and groundwater containing different ion exchange membranes and a buffering zone. This BES is based a modified microbial desalination cell (MDC);8,9 although its structure and operation assembles electrodialysis (ED10), the BES is different from an ED because the electron source in an BES is organic compounds and electrons are generated from organic oxidation catalyzed by microorganisms;11 while the electrons in an ED are from water electrolysis. Laboratory investigation was conducted with four objectives: (1) to demonstrate the feasibility of this BES for nitrate removal; (2) to understand the competition of anion transport during nitrate removal; (3) to investigate water consumption by nitrate removal; and (4) to accomplish simultaneous nitrate reduction.
2 Material and methods
2.1 BES construction and operation
The BES was constructed similarly to a tubular microbial desalination cell,12 consisting of two membrane tubes: the inner tube made of a piece of cation exchange membrane (CEM, Membrane International, NJ, USA) as an anode chamber (with a liquid volume of 500 mL), and the outer tube made of a piece of anion exchange membrane (AEM, Membrane International, NJ, USA). The space between the two tubes created a concentrating chamber (∼250 mL), designed to accumulate nitrate ions extracted from groundwater. The BES did not have a “cathode chamber”; when it is submerged in groundwater, the water well would act as a “cathode chamber”. In this study, the BES was placed into a 2 L tank containing synthetic groundwater (deionized water and sodium nitrate). The anode electrode was a 20 cm long carbon brush inserted into the anode chamber with a rubber spiral spacer as described previously.13 The cathode electrode was a piece of carbon cloth coated with 0.5 mg Pt cm−2 that wrapped the outer AEM tube.During electricity generation, ionic transport accompanies electron transfer: when electrons move from the anode electrode to the cathode electrode via the external circuit, cations will move from the anode chamber into the concentrating chamber through CEM, while anions (including nitrate and other anions present in groundwater) will migrate across AEM from groundwater into the concentrating chamber (Fig. 1). In this way, nitrate is separated from groundwater without introducing any ions from the anolyte (because of the barriers of the concentrating chamber and the different ion exchange membranes).
 | ||
| Fig. 1 Schematic and experimental setup of the BES used in this study for nitrate removal. | ||
The BES was operated at a room temperature of ∼20 °C. The anode was inoculated with anaerobic sludge from a local wastewater treatment plant (South Shore, Milwaukee, WI, USA), and was continuously fed with a solution containing (per liter of tap water): CH3COONa, 0.5 g; K2HPO4, 1.07 g; KH2PO4, 0.53 g; NH4Cl, 0.075 g; NaCl, 0.25 g; MgSO4, 0.0075 g; CaCl2, 0.01 g; NaHCO3, 0.05 g; trace mineral solution 0.1 mL;14 and yeast extract, 0.1 g. The anolyte was fed at a flow rate of 0.7 mL min−1 and was recirculated at 100 mL min−1. The concentrating chamber was supplied with deionized water (containing no nitrate) at the designated flow rates. The synthetic groundwater was prepared by dissolving NaNO3 in deionized water and was replaced with fresh medium at the end of each cycle; its conductivity was maintained at about 200 μS cm−1. The BES was operated with or without 0.8 V externally applied through a circuit as introduced before.7
2.2 Measurement and analysis
The BES voltage was recorded every 5 minutes by a digital multimeter (2700, Keithley Instruments, Inc., Cleveland, OH, USA). The pH of the anolyte, the solution in the concentrating chamber, the synthetic groundwater was measured by a benchtop pH meter (Oakton Instruments, Vernon Hills, IL, USA). The conductivity was measured by a benchtop conductivity meter (Mettler-Toledo, Columbus, OH, USA). The concentrations of chemical oxygen demand (COD) and nitrate–nitrogen (NO3−–N) were measured by using a colorimeter following the manufacturer's instructions (Hach DR/890, Hach Company, Loveland, CO, USA). Charge transfer efficiency was calculated as previously described:15where, Qth is the theoretical amount of coulombs required to move nitrate ions that are removed within an operating cycle (assuming one electron is needed to move one nitrate ion), and Q stands for coulombs harvested from electric current during an operating cycle.

3 Results and discussions
3.1 BES Feasibility
The BES was started in a microbial fuel cell (MFC) mode without additional voltage applied but an aerated cathode for acclimation of the anode microbial community. After two-week MFC startup, an external voltage of 0.8 V was applied to the circuit and thus the BES was turned to an MEC (microbial electrolysis cell) operation. The concentrating chamber was supplied with DI water and refilled at the end of each cycle during the start-up stage, and then switched to a continuous mode at flow rate of 0.7 mL min−1, resulting in an HRT of 6 h. With an input COD of ∼450 mg L−1, the anode of the BES removed more than 70% of organic compounds at an HRT of 12 h. The BES achieved an average current density of 28 A m−3 upon replacement of the synthetic groundwater (Fig. 2A).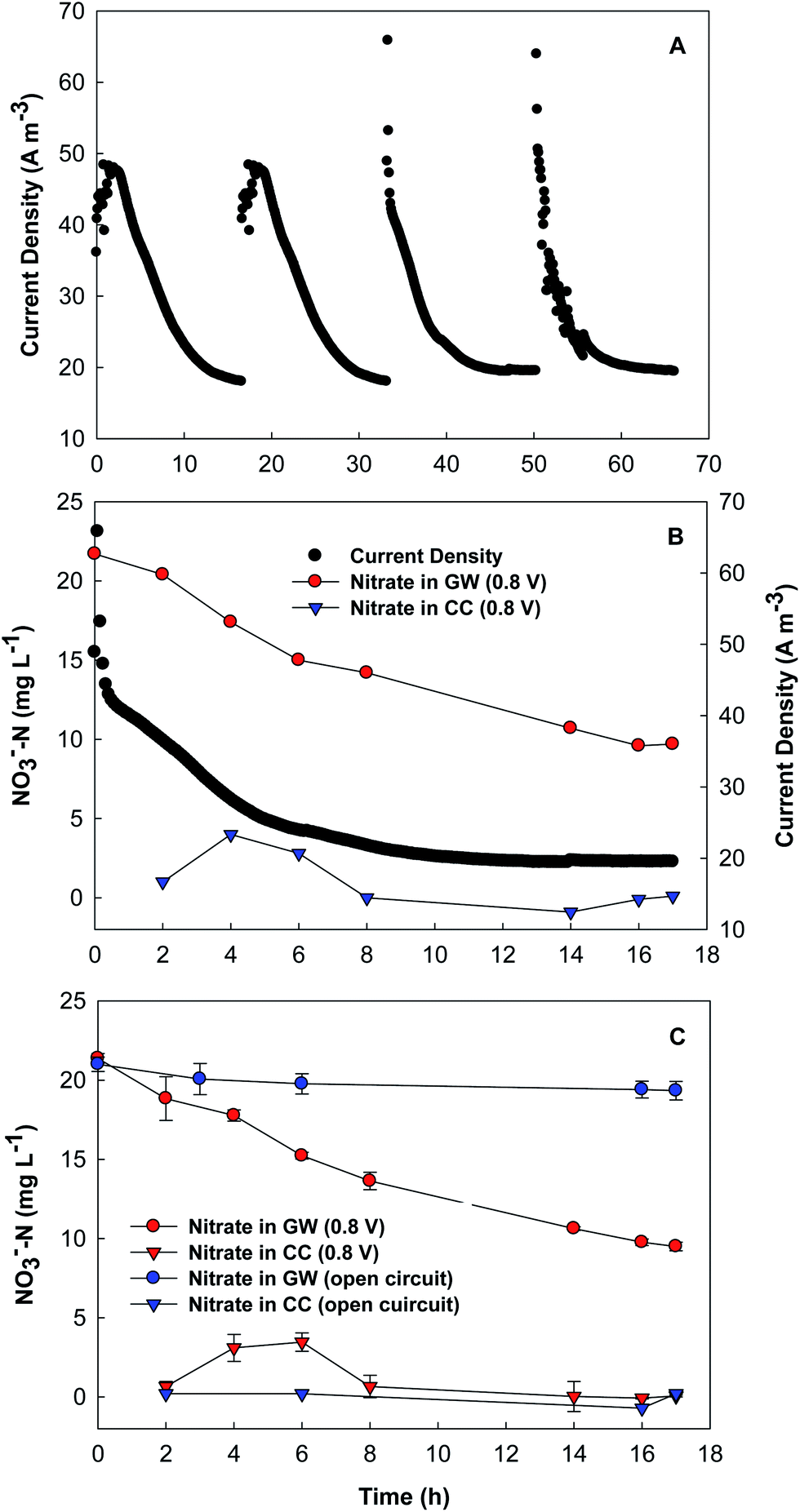 | ||
| Fig. 2 The BES performance: (A) current generation; (B) the correlation between nitrate concentration and current generation; and (C) the nitrate concentrations under the open – and the closed – circuit conditions. GW: groundwater; CC: concentrating chamber. | ||
The current profile seemed to be consistent with the variation of nitrate concentration in the synthetic groundwater (Fig. 2B). One can see that the nitrate concentration decreased from 21.4 ± 0.2 to 9.5 ± 0.3 mg L−1 within a batch of 17 h, while current density decreased from ∼40 to ∼20 A m−3. In the first 6 h of a batch, nitrate was accumulated in the concentrating chamber, demonstrating that nitrate migrated away from the synthetic groundwater, and then its concentration started to decrease, likely due to the decreased electric current (and thus less nitrate movement) and strong dilution effect because of the continuous flow of DI water in the concentrating chamber. When the supply of DI water into the concentrating chamber was changed to a batch mode for a short period of time, we were able to quantify the mass of nitrate nitrogen and found that the total amount of nitrate nitrogen in the concentrating chamber was 18.9 ± 0.6 mg, which matched the loss of nitrate nitrogen from the synthetic groundwater (18.1 ± 0.3 mg) during the same time period. This result indicates that nitrate was not removed via biological reduction on the cathode (either bioelectrochemical denitrification using the cathode electrode as an electron donor, or autotrophic denitrification using hydrogen gas as an electron donor), which was not expected in the absence of microorganisms.
To confirm the role of electrical current in driving nitrate ions movement, the BES was operated under an open-circuit condition for comparison. As shown in Fig. 2C, the nitrate concentration in the synthetic groundwater slightly decreased (by about 2 mg L−1) under the open-circuit condition, and no nitrate was detected in the concentrating chamber. The obvious difference between the open- and the close-circuit conditions demonstrates that current generation is an indispensable factor for nitrate removal from groundwater by using the present BES.
3.2 Competition for anion transport
We observed that the nitrate concentration in the synthetic groundwater remained at 9–10 mg L−1 after 17 h operation and did not further decrease. It seems that there was a balance of nitrate ions between the synthetic groundwater and the concentrating chamber. To further understand this phenomenon, we examined the BES performance with 50 mg L−1 as an initial nitrate concentration in the synthetic groundwater. The BES generated an average current density of 29.6 ± 3.4 A m−3, and it took more than 20 h for current generation to reach stable, likely due to a higher initial nitrate concentration (more nitrate ions for being removed). The nitrate concentration in the synthetic groundwater decreased to around 20 mg L−1 and no further decrease was observed. This result shows that the BES did reach equilibrium of nitrate concentration and the final nitrate concentration was affected by the initial nitrate concentration. The charge transfer efficiency with an initial nitrate concentration of 20 mg L−1 is 25.1 ± 6.3%, suggesting that the charge associated with nitrate ion migration was much smaller than the charge generated from electric current and thus there must be other anion species moving through AEM to meet the requirement of charge transfer by current generation. A potential candidate of the additional anion is OH−, generated from proton reduction on the cathode electrode (considering that other ions in the synthetic groundwater are in trace amount).To verify this, we changed the feeding of the concentrating chamber from continuous flow to batch operation (with internal circulation) so that the accumulation of OH− in the concentrating chamber might be detected by measuring the pH (a continuous feeding would dilute the pH effect). Because of bioelectrochemical reactions, H+ in the groundwater was reduced to hydrogen gas with the accumulation of OH−, which raised the pH of the synthetic groundwater from 6.6 ± 0.5 to 10.7 ± 0.1. We do not expect that the deployment of BES for in situ groundwater remediation will significantly change the pH of groundwater, because of a large quantity of groundwater and the associated dilution effect; however, the pH in the local area adjacent the BES may increase. To facilitate current generation, ions need to move from the anode and the cathode into the concentrating chamber (to maintain a charge balance in all three chambers). In the synthetic groundwater, the anions present in a large quantity are nitrate and OH− ions, and the migration of OH− into the concentrating chamber increased its pH to 12.0 ± 0.2. The amount of OH− in the concentrating chamber was about 2.5 × 10−3 mol, higher than the amount of nitrate ions moving in (1.3 × 10−3 ± 2.1 × 10−5 mol). Therefore, OH− ions are a strong competitor with nitrate ions during anion migration, and it is likely that the transport of OH− ions inhibited nitrate ions movement to a certain extent (and eventually nitrate movement was significantly slowed down), because of a higher concentration gradient, smaller molecule, and/or stronger bonding with the charge group of AEM.16–18
3.3 Water consumption by nitrate removal
At a flow rate of 0.7 mL min−1 (HRT of 6 h), removing 1 mg NO3−–N from the synthetic groundwater would require more than 30 mL DI water. This water consumption could be significant due to a large quantity of nitrate ions in groundwater. To investigate whether water consumption could be reduced, we examined the BES performance with two smaller flow rates, 0.2 and 0.1 mL min−1, corresponding to the HRT of 20 and 42 h. The total Coulomb produced in a cycle was 1047.4 ± 105.2 C at the HRT 42 h, followed by 854.5 ± 75.5 C at 6 h and 752.2 ± 50.1 C at 20 h (Fig. 3A), indicating that reducing the supply of DI water to the concentrating chamber did not decrease electricity generation. Water consumption per mg nitrate removal was drastically decreased by lowering the flow rate: removing 1 mg of nitrate nitrogen would need 11.7 ± 1.6 and 5.2 ± 0.39 mL DI water at the HRT of 20 and 42 h, much less than 35.9 ± 3.6 mL at 6 h (Fig. 3B). The pH of the water in the concentrating chamber increased with the smaller flowrate (Fig. 3C), likely because of less dilution effect; while the pH of the synthetic groundwater was similar among the three HRTs (Fig. 3D). Although less DI water supply (e.g., at the HRT of 42 h) could reduce operating cost, it accumulated higher concentrations of both nitrate ions (12.5 ± 1.0 mg NO3−–N L−1) and OH− (a higher pH), compared with more water supply (10.1 ± 0.4 mg NO3−–N L−1 and a lower pH at the HRT of 6 h), which could create a larger concentration gradient against further migration of nitrate ions from groundwater. The effect of such an increased concentration gradient on nitrate removal warrants further investigation.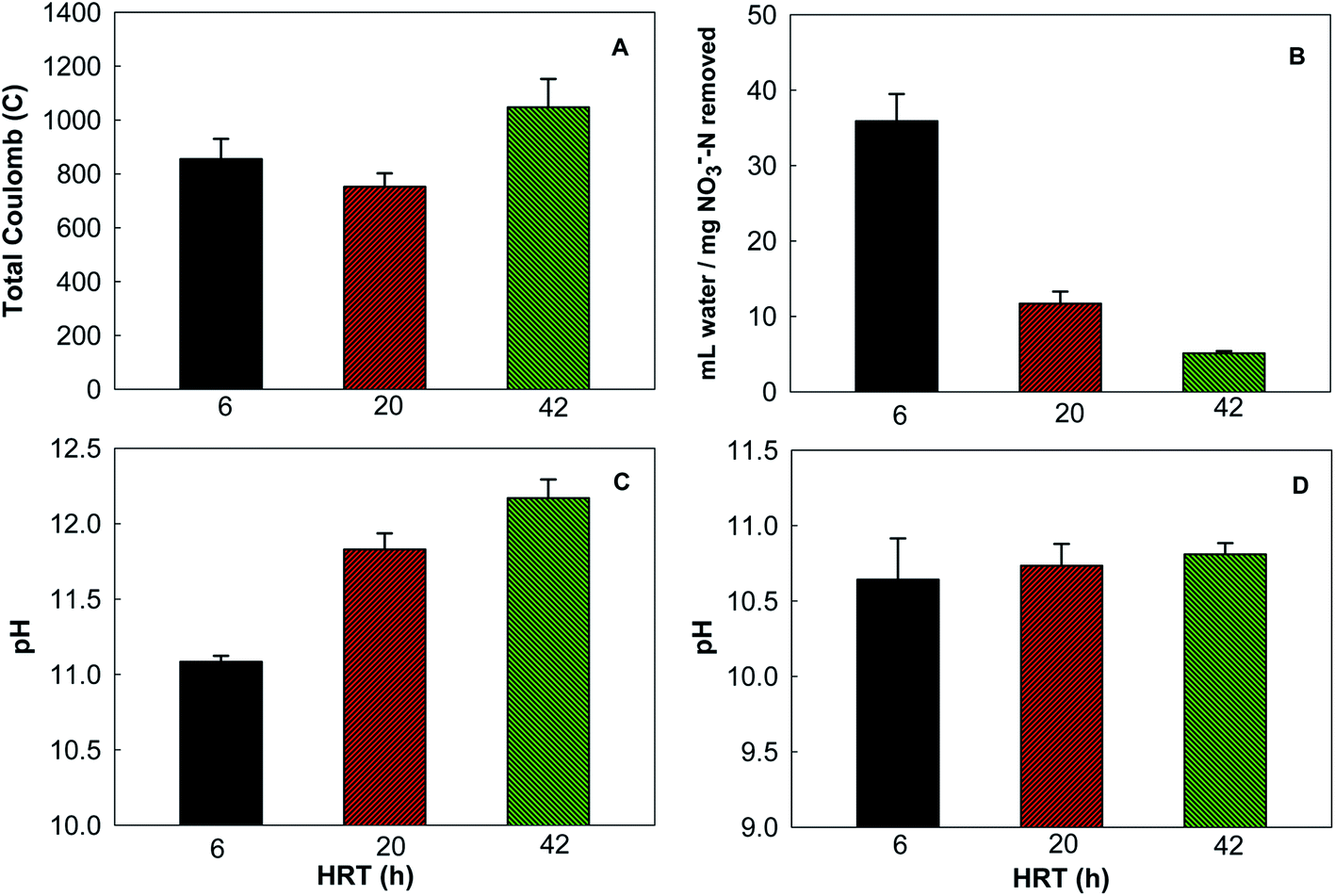 | ||
| Fig. 3 The results of varying the flow rate (or HRT as shown by the x-axis) of the DI water supply to the concentrating chamber: (A) total Coulomb production; (B) the water consumption per mg nitrate nitrogen removed; (C) the pH of the water in the concentrating chamber; and (D) the pH of the synthetic groundwater. | ||
3.4 Nitrate reduction in the anode
In the present BES, nitrate was accumulated in the concentrating chamber, rather than being reduced to nitrogen gas. Further action would be needed to “completely” remove nitrate. To realize nitrate reduction, we transferred the effluent of the concentrating chamber into the anode chamber at a flow rate of 0.1 mL min−1; the flow rate of the mixed anode feeding solution was at 0.7 mL min−1. Within a cycle of 17 h, there was 10.4 ± 0.3 mg of nitrate nitrogen flowing into the anode chamber accompanied with 271 ± 43.8 mg COD consumed. The average current density was 33.6 ± 0.1 A m−3. We did not detect any nitrate in the anode effluent, and the nitrite nitrogen was below 0.3 mg L−1, suggesting that nitrate was effectively reduced.3.5 Perspectives
The present BES has several potential advantages: (1) the in situ treatment avoids the energy-intensive pump-and-treat approaches; although the supply of the anolyte will require some pump energy, it should be much less than that of pumping groundwater (which has a vast volume); (2) extracting nitrate out of groundwater avoids the addition of chemicals and the stimulation of bacterial growth like that in other in situ chemical or bioremediation; and (3) multiple barriers (consisting of two different ion exchange membranes and one concentrating chamber) avoids the intrusion of undesired ions and microorganisms moving from the anolyte into groundwater and thus allows the use of low-grade electron donors such as wastewater.This study provides a proof of concept, and thus we must also understand the challenges and issues that should be addressed in the future study: (1) the use of DI water in the concentrating chamber will not be practical, and although its volume could be significantly reduced, during practical application a large amount of DI water will still be needed. The possible solution to address this problem is to use groundwater or treated anolyte; (2) the energy consumption by this process should be analyzed; the energy is input in two ways, direct electric potential application by the power supply and energy to drive the pumping systems to transport water; (3) the pH change in groundwater needs more investigation, and we need to understand how extensively this pH effect will be (e.g., how serious the deposition of hardness ions on membranes will be); (4) the effectiveness of the BES needs to be demonstrated with actual groundwater and wastewater; and (5) further scaling up of the BES is always a challenge. For in situ deployment in water wells, the present BES may be developed to a scale of several hundred liters.
Acknowledgements
This study was financially supported by a grant of Research Growth Initiative (RGI) from University of Wisconsin-Milwaukee.References
- J. O. Lundberg, E. Weitzberg, J. A. Cole and N. Benjamin, Nat. Rev. Microbiol., 2004, 2, 593–602 CrossRef CAS PubMed
.
- C. D. Rocca, V. Belgiorno and S. Meriç, Desalination, 2007, 204, 46–62 CrossRef PubMed
- H. Wang and Z. J. Ren, Biotechnol. Adv., 2013, 31, 1796–1807 CrossRef CAS PubMed
- W.-W. Li, H.-q. Yu and Z. He, Energy Environ. Sci., 2014 10.1039/c3ee43106a
- P. Kelly and Z. He, Bioresour. Technol., 2014, 153, 351–360 CrossRef CAS PubMed
- Y. Zhang and I. Angelidaki, Water Res., 2013, 47, 1827–1836 CrossRef CAS PubMed
- Y. Tong and Z. He, J. Hazard. Mater., 2013, 262, 614–619 CrossRef CAS PubMed
- X. Cao, X. Huang, P. Liang, K. Xiao, Y. Zhou, X. Zhang and B. E. Logan, Environ. Sci. Technol., 2009, 43, 7148–7152 CrossRef CAS
- B. Zhang and Z. He, RSC Adv., 2012, 2, 10673–10679 RSC
- V. K. Gupta, I. Ali, T. A. Saleh, A. Nayak and S. Agarwal, RSC Adv., 2012, 2, 6380–6388 RSC
- D. Pant, A. Singh, G. V. Bogaert, S. I. Olsen, P. S. Nigam, L. Diels and K. Vanbroekhoven, RSC Adv., 2012, 2, 1248–1263 RSC
- K. S. Jacobson, D. Drew and Z. He, Environ. Sci. Technol., 2011, 45, 4652–4657 CrossRef CAS PubMed
- F. Zhang, Z. Ge, J. Grimaud, J. Hurst and Z. He, Bioresour. Technol., 2013, 134, 251–256 CrossRef CAS PubMed
- L. T. Angenent and S. Sung, Water Res., 2001, 35, 1739–1747 CrossRef CAS
- X. Chen, X. Xia, P. Liang, X. Cao, H. Sun and X. Huang, Environ. Sci. Technol., 2011, 45, 2465–2470 CrossRef CAS PubMed
- T. H. J. A. Sleutels, H. V. M. Hamelers, R. A. Rozendal and C. J. N. Buisman, Int. J. Hydrogen Energy, 2009, 34, 3612–3620 CrossRef CAS PubMed
- Y. Fan, E. Sharbrough and H. Liu, Environ. Sci. Technol., 2008, 42, 8101–8107 CrossRef CAS
- P. Długołęcki, K. Nymeijer, S. Metz and M. Wessling, J. Membr. Sci., 2008, 319, 214–222 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2014 |