Engineered fluorescence tags for in vivo protein labelling
Zhipeng Wangabc,
Xiaozhe Dingb,
Sijian Lia,
Jing Shid and
Yiming Li*ab
aSchool of Medical Engineering, Hefei University of Technology, Hefei, Anhui 230009, China. E-mail: lym2007@mail.ustc.edu.cn
bDepartment of Chemistry, School of Life Sciences, Tsinghua University, Beijing 100084, China
cDepartment of Chemistry, Princeton University, Princeton, New Jersey 08544, USA
dDepartment of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 7th January 2014
Abstract
In vivo protein labelling with a peptide tag–fluorescent probe system is an important chemical biology strategy for studying protein distribution, interaction and function. A variety of engineered tags have been developed to label proteins of interest (POIs), in which a protein or peptide tag is covalently linked to the POIs that can afford fluorescence signal under certain conditions. This review summarizes the current methodology in five categories, namely autofluorescent protein tags, peptide tags with a specific probe-binding amino acid sequence, peptide tags that can be further labelled by enzyme recognition, peptide tags that contain an enzymatic domain which can interact with small molecules or coenzymes, and peptide tags that can be labelled with probes through natural or artificial protein–protein interaction. The principles and application of each tool are also reviewed.
1. Introduction
Proteins play fundamental roles in most life processes. Therefore, there is a strong need to study the structures, locations, functions and enzyme mechanisms of them per se with exact spatiotemporal resolutions to establish a clear picture under physiological conditions.1 Large numbers of diverse methods have been developed for protein tracking, such as radioactive labeling, isotope markers, and electrochemical probes.2 Fluorescent labeling is regarded as one of the most common and versatile weapons among them. Besides routinely used fluorescent proteins, the last decade has witnessed the development of many in vivo labelling methods.3 In these methods, a special optimized or designed peptide or protein tag is genetically fused to POIs, followed by some orthogonal labelling strategies. Some of the probes are able to penetrate the cell membrane and label proteins in cytoplasm; while some can only be used for labelling membrane proteins.4Diverse and colorful principles filled up the labelling arsenal, and they can be roughly divided into the following five categories: autofluorescent protein tags, peptide tags with specific probe-binding sequence, peptide tags labelled by specific enzyme recognition, peptide tags containing enzymatic domain for interacting with small molecules or coenzymes, and peptide tags labelled with probes through natural or artificial protein–protein interaction. The present manuscript aims to review the concepts, recent developments, and applications of these categories of engineered fluorescence tags for in vivo protein labelling. With these living cell labelling methods in hand, many significant biological questions, which have been difficult to solve for a long period, should have clearer explanations. Besides labelling POIs with chromophores, selective protein modification can also provide a path to the formation of polymer–protein conjugations, protein post-translational modifications, antibody–drug conjugates or linkage with any small molecule from chemical synthesis.5
Besides the methods listed above, unnatural amino acid incorporation method (UAA) developed by Schultz and co-workers6 can also be used to label POIs. In this method, amino acids with diverse side chains can be introduced into POIs for fluorescent imaging or interacting with other probes by bio-orthogonal reaction.7 Because we are only focusing on tags that are genetically fused to terminal parts of POIs in this review, the UAA incorporation method will not be discussed although it is also a widely accepted strategy for labelling via genetic engineering.
2. Autofluorescent protein tags
To begin with, it would be simple and convenient if the genetically encoded tag can emit fluorescence itself, which means that no more subsequent labelling steps are needed. The development of fluorescent proteins (FPs) like Green Fluorescent Protein (GFP) and their variants enabled the application of this kind of tags. When exposed to light range from blue to UV, GFP emits green light without using any substrates or coenzymes.8 GFP, which contains 238 amino acids, was originally discovered in jellyfish Aequorea victoria.9 Functional GFP was then successfully cloned10 and expressed11 in other organisms.The principle of autofluorescent for this protein family is the formation of 4-p-hydroxybene-5-imidazolinone core structure by the side chains of Serine 65, dehydro-Tyrosine 66 and Glycine 67 (Fig. 1). Thus, one of the primary advantages of fluorescent protein tags is that the formation of chromophore is completely genetically encoded, therefore the chromophore is directly fused to POIs and no extra cofactor is needed. This advantage makes the targeting process simple and precise. In addition, as a natural protein, FPs show less photo-toxicity compared to simple chemical dyes. Although one molecule of H2O2 is generated during the formation of each chromophore8 which in theory could be potentially toxic, it seems that cells have detoxicating mechanisms against this by-product and therefore little reactive oxygen species is actually produced.12
 | ||
| Fig. 1 Chromophores of some major FPs used as fluorescent tags with various exciting and emitting spectra. | ||
2.1 Strengthened GFP
Based on wild-type GFP, a large number of its variants have been evolved through protein engineering and result in a rich variety of tools for protein labelling. Much work has further been focused on improving fluorescent protein folding and expression properties. One of the most widely used GFP variant named Enhanced GFP (EGFP) combines a set of optimizations and mutations that increase expression yields of mammalian cells, brightness and stability of GFP. These optimized properties13 allow visualization of POIs for hours with only little photobleaching, or even quantitatively. Another improved version of EGFP, superfolder GFP, was made by adding a “cycle-3” mutation and other six new mutations on EGFP.14 This variant can fold well even when fused to unfolded polypeptides, which makes it a useful weapon with special applications.15,162.2 FPs with expanded fluorescent spectra
Excitation and emission spectra range of FPs have been expanded tremendously since the discovery of GFP. By mutating GFP, Tsien's group first engineered blue fluorescent protein (BFP) and cyan fluorescent protein (CFP),17 while yellow fluorescent protein (YFP) was later rationally designed based on the three dimensional structure of GFP.18 Fluorescent proteins with red-shifted excitation and emitting spectra can act well as superior tags for in vivo imaging because of stronger penetration ability, lower autofluorescence background, less light-scattering, and less phototoxicity.19 However, the initial attempts to engineer variants of Aequorea GFP with red-shifted emission maximum longer than 529 nm did not succeed until 2008.20 Instead, early red-shifted fluorescent proteins (RFPs) were found in nature. Lukyanov group21 reported first RFPs discovered in Anthozoan animals in 1999. DsRed, one of the most widely used RFP, was later discovered in Discosoma.22,23 The diversity of fluorescent protein spectra allows multicolor imaging and various choices of FRET pairs.It should be mentioned that some FPs have changeable fluorescence, which are conveniently used for tracing of various biological events and signals. For example, destabilized GFP can be used to detect dynamic cellular activity.24
The fluorescence intensity of engineered FPs which can respond to certain signals or conditions, or smart labels, are also widely used.25
In spite of the positive properties and promising applications of autofluorescent protein tags, several limitations do exist. Firstly, the large size of FP tags might have steric hindrance effect on POIs or even prevent normal cellular activities. For instance, YFP tag may interfere with translocation of POIs into the nucleus.26 Secondly, introduction of FP tags needs ectopic expression, which may also perturb POIs' normal structure and function. Moreover, another limitation which should be emphasized is that autofluorescent protein tags can only be fused to the N- or C-terminal of POIs.
3. Peptide tags with specific probe-binding sequence
Theoretically, a peptide tag with any amino acid sequence can be easily fused to the terminals of POIs via routine protein engineering strategy. The combinations of different side chain function groups enable diverse physical or chemical interactions with ligands. Therefore, a sophisticatedly designed sequence could be used for fluorescence labelling.3.1 Biarsenical ligands
Tsien group27 developed a pioneering method for selectively protein labelling. In this method, biarsenical ligands (such as FlAsH–EDT2) serve as probes that can specifically bind to proteins with four cysteines at i, i + 1, i + 4, i + 5 sites of the α-helix structure. This peptide tag with a tetracysteine motif (CCXXCC sequence or TC sequence, X means non-cysteine amino acid) can be linked to recombinant POIs via genetic fusion or insertion (Fig. 2). Because of the high permeability and small size of biarsenical fluorescence probes, they can easily penetrate cell membrane without disturbing cell metabolism. Abovementioned advantages should facilitate the usage of such strategy both in vitro and in vivo.28 Compared with the unbound sample, the sample selectively bound to TC sequence produces a much higher fluorescence signal. Tsien group29 further improved the binding kinetics by optimizing the sequence. The preferred sequence CCPGCC turned out to have a hairpin structure rather than previous reported α-helix. A series of FlAsH derivatives, such as ReAsH–EDT, were synthesized to own different colors or usages.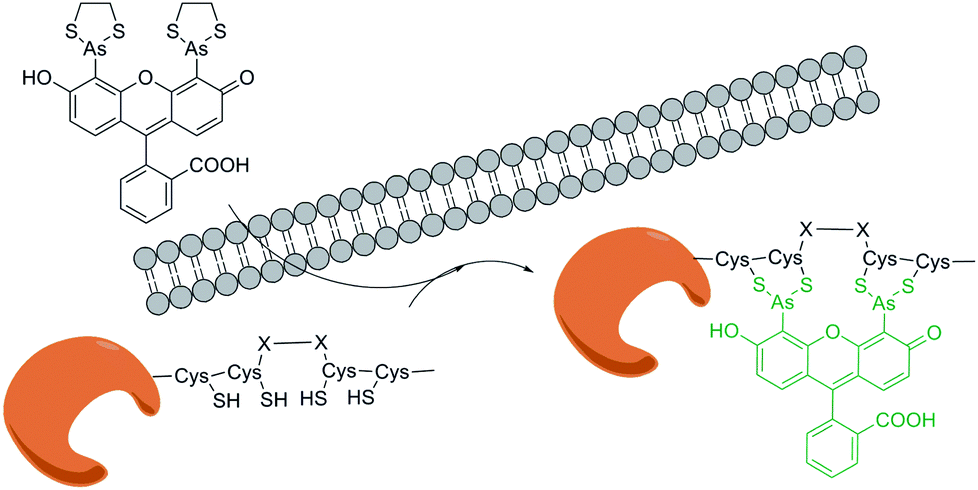 | ||
| Fig. 2 Protein labelling by tetra-cysteine tag interaction with biarsenical ligands. | ||
Noting that the biarsenical ligands can bind to the outer face or the inner structure without interfering normal functions of the protein, this technique has been used in diverse systems. For example, Tsien group30 studied the conformational change of GPCR in the activation process by measuring FRET between biarsenical ligand and CFP tag. Recently, Orner group31 developed a method to detect protein monomers forming cage-like oligomer structures. The strategy is to engineer protein monomers with cysteine pairs presented near each other when they assemble as cage state. FlAsH can then bind to the assembled pairs of cysteines in the cage state. It should be emphasized that the advantage of this method is owe to its capability of labelling proteins intracellularly.
Although these biarsenical ligands have been applied widely, they still suffer from high toxicity, noisy background, as well as limited usage in cellular locales with oxidative environment. To overcome these limitations, Schepartz and co-workers32 developed boronic acid based ligands (RhoBo) that can similarly bind to tetraserine motifs and label the POIs. Besides high selectivity, convenience and brightness, this tag family owns additional advantages such as nontoxicity and redox-insensitivity.
3.2 His tag–Ni(II) ion coordination probe
His-tag has been used as one of the classical specific tag methods for protein purification, functionalization and immobilization.33 The fundamental principle of this process is the high affinity binding between the oligo-histidine sequence and the nickel ion, which is first discovered by Hochuli group in 1987.34 In 2004, Vogel group35 described another useful labelling strategy for membrane proteins mostly based on this simple principle of affinity binding: genetically encoding His-tag binds with complexes such as Ni(II) nitrilotriacetic acid (Ni II–NTA) with a fluorescent probe (Fig. 3). | ||
| Fig. 3 Protein labelling by histidine tag interaction with Ni(II) complex. | ||
One year later, Lippard and co-workers36 introduced a new probe system, which could overcome shortcomings such as pH-sensitivity and easily quenching properties as previously reported. The fluorescent dye NTA–DCF can coordinate with Ni(II) with only a little impact on the emission of dyes per se. By comparing RFP with the RFP–His6 transfected, Lippard group confirmed the accuracy and selectivity for labelling the His tag on extracellular POIs, and proposed its potential application in intracellular POI labelling. Compared with probes covalently bound with a single function group, it is worth mentioning that tags based on coordinate binding of probes with target peptides instead of a single amino acid residue could result in improved selectivity.
3.3 Asp tag–Zn(II) ion coordination probe
Based on similar ideas, people can utilize the coordination reaction between other ions and the side chain of certain amino acids. Hamachi and collaborators37 discovered that oligo-aspartate peptide sequence, termed D4 tag, can bind to multinuclear Zn(II) complexes such as Zn(II)–DpaTyrs (Fig. 4). Because of the multiple coordination bonds between the carboxyl group and the ion, as well as the multivalent effect, the binding shows very high stability. Thus, the POIs will be labelled by fluorescence if the Zn(II) complexes carry a chromophore. This design was first achieved in test tubes, and then membrane protein was labelled by fusing with fluorescent (D4)n (n range from 1 to 3) probes. The results demonstrated that very few interference on its natural biological function was detected by using abovementioned system.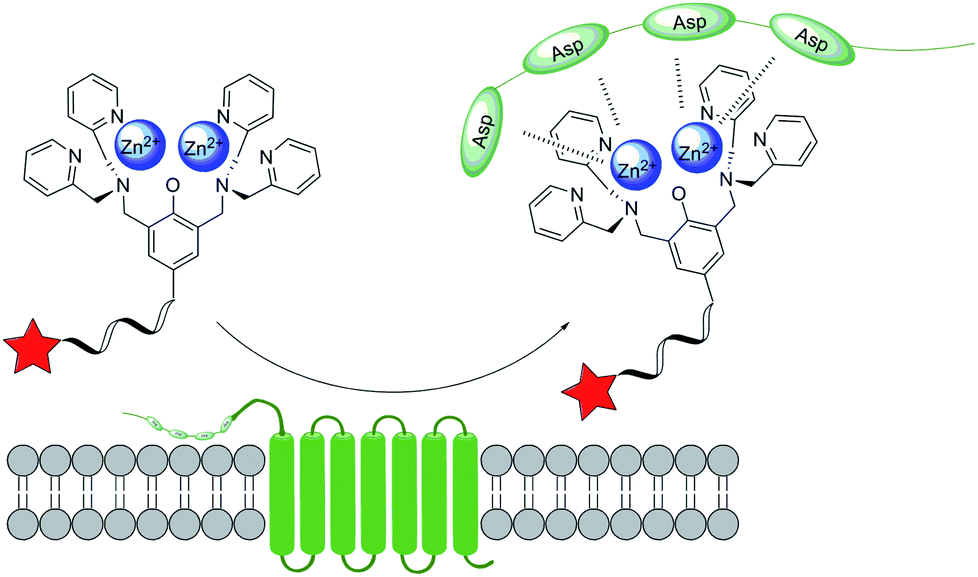 | ||
| Fig. 4 Protein labelling by aspartic tag interaction with Zn(II) complex. | ||
Subsequently, they38 designed and developed a POIs' detection system based on the same labelling method. Additionally, seminaphthorhodafluor (SNARF) can show the change of the pH in the microenvironment after binding. Thus, SNARF linked with Zn(II)–DpaTyr performed well as a ratiometric probe for membrane proteins in vivo or POIs in vitro. In 2009, Hamachi et al.39 expanded the usage of this novel tag to the study of G-protein coupled receptor proteins (GPCR), demonstrating that this method act as a powerful weapon for studying membrane proteins. This study indicated that CA6D4x2 tag (a certain kind of D4 tag with a sequence of CAAAAAADDDDGDDDD) can selectively and rapidly bind with a tetranuclear Zn(II)–DpaTyr probe. This work is the first example for realizing non-enzymatic labelling of membrane protein with small molecule.
In 2010, Hamachi and co-workers40 screened a variety of metal ions as well as peptide sequences for optimizing binding pairs. They found that Ni(II)–DpaTyr could bind to a multiple D3 tag peptide, such as DDDXXDDD sequence, with high affinity. Furthermore, they measured the binding affinity and proposed its application in β-galactosidase inactivation study. They41 used Zn(II) as a surrogate of Ni(II) in this multiple D3 tag, which could avoid fluorescence quenching and toxicity caused by the introduction of nickel. Moreover, Zn(II)–Ida was developed as the novel fluorescent probe for labelling GPCR with a His-tag in vivo. It is worth to mention that Tsien and his collaborators42 also developed a fluorescent Zn(II) complex for selectively binding with His-tag. Compared with the former ones, this probe exhibited a complete different structure. This application was further expanded by labelling of membrane protein STIM1 fused with hexa-histidine sequence.
3.4 Bio-orthogonal reaction based labelling system
Combined with bio-orthogonal reactions, a peptide tag can be selectively modified on a certain amino acid residue, and thus POIs with the peptide tag are labeled. Francis group43 discovered a special Trp modification method with metallocarbenes under wide pH conditions. Additionally, they showed that a highly solvent-accessible 26-peptide melittin can serve as a peptide tag for targeted labelling, i.e. FKBP–melittin. This method can be traced back to 2004 when Francis and the collaborators44 labeled subtilisin and myoglobin via dirhodium tetraacetate catalyzing tryptophan modification. Despite these important advances, the practical application of this labelling method is still limited. Till now, this system is only applicable for tryptophan and a large amount of rhodium compound is needed for the labelling process.454. Peptide tags labelled by specific enzyme recognition
One of the exquisite characteristics of natural enzymes whose substrates are peptides or proteins is that their recognition of substrates is highly specific. Therefore, the whole protein should be selectively realized by an enzyme when fused with a sequence that acts as the substrate of the enzyme. Based on this principle, target POIs could be labelled with small fluorescent molecules by enzyme catalyzing reaction.4.1 LplA–LAP system
Recently, Ting group46 developed a novel small fluorescent molecule labelling technique named PRobe Incorporation Mediated by Enzymes (or PRIME). The basic biological foundation of this technique is that the native lipoic acid ligase (LplA) from Escherichia coli can add a small molecule to LplA Acceptor Peptide (LAP) containing only 13 amino acids. Through protein mutagenesis and molecular evolution, they optimized LplA and got some new enzymes which can recognize analogs of the natural small molecule substrate. Generally, the labelling process with LplA–LAP system can be divided into two categories: directly ligating a small fluorescent molecule with the peptide tag or two-step labelling combining bio-orthogonal reactions. Ting and co-workers47 carefully studied the mechanism and applications of the recognition of 7-hydroxycoumarin by mutant LplA enzyme system, which belongs to the this category. Another molecule 7-aminocoumarin was also verified as a protein labelling tag.48On the other hand, making use of myriad bio-orthogonal reaction can vastly extend the arsenal. If a trans-cyclooctene based molecule is ligated to the tag, a tetrazine probe can label the POIs by Diels–Alder reaction.49 Moreover, strain-promoted click reaction50 between an internal copper-chelating segment with a normal alkyne,51 and the hydrazone formation reaction between an aldehyde and a labelled hydrazine moiety or vice versa52 were reported. It should be mentioned that through an optimized cyclooctyne structure and washing step to remove the excess probes, the strain-promoted click reaction labelling method can be used for POIs inside living cells. Combined with HaloTag strategy, a two-step fluorescent probe53 and quantum dot (QD)54 labelling method were developed.
At the same time, this system can be further applied in several other applications. With an aryl–azide derivate serving as a photo-cross-linker, the system can be used to detect protein–protein interaction (Fig. 5).55
 | ||
| Fig. 5 Protein labelling by different LplA–LAP systems. | ||
4.2 Transglutaminase–Q tag system
Transglutaminase (TGase) can catalyze the reaction between the side chains of Gln and Lys to form an isopeptide bond. Because it has a high tolerance over different amino group moieties but is specific on Gln's carboxyl group part, other amino group containing probes could also replace Lys and act as substrates, such as biotin–cadaverine and fluorescein–cadaverine.56 Three diverse Q tags with different sequences (Q1, Q2 and Q3) were developed which can be recognized by transglutaminase and further linked to many functionalized probes.574.3 Biotin ligase–AP systems
Biotin ligase BirA from Escherichia coli can selectively biotinylate the lysine side chain amino group of an acceptor peptide (AP or Avitag) with 15 amino acid. Therefore, AP can perform as a fusing tag for membrane POIs and be labelled by biotin. Due to the high affinity of the interaction between biotin and streptavidin, QDs can connect with cell surface protein to label the membrane POIs for further biological studies.58 A complete protocol of this method has already been presented59 and various applications such as an assay to detect receptor oligomerization state have been reported.604.4 APEX systems
Ting group61 also developed an electron microscopy assistant tag enzyme linked with POIs termed “APEX”. In this strategy, the in situ polymerization of 3,3′-diaminobenzidine (DAB) catalyzed by APEX can be subsequently stained by OsO4, which was able to increase the contrast for EM. Recently, Ting and the collaborator have already reviewed other POIs in vivo labelling methods developed by their group.624.5 Other similar systems
Several other groups have also contributed to this subfield through developing diverse enzyme systems. Bertozzi and co-workers63 introduced a 6-amino-acid tag system with a sequence of LCTPSR. The cysteine can be recognized and modified with formylglycine (fGly) by formylglycine-generating enzyme (FGE). Because the formyl group can be linked to fluorophores via imine bond, this technology is termed “aldehyde tag”. In the following researches, they64 extended the usage for this method to form different chemical conjugations with diverse cargoes. An important transpeptidase sortase has also been developed for protein labelling. Sortase can cleave the amide bond between Thr and Gly in the peptide tag LPXTG, and subsequently catalyze its formation of new bonds with peptide or peptide conjugation moiety containing oligoglycine. Based on this principle, POIs could be labelled with biotin or fluorescence probes,65 and this strategy was named as sortagging. For example, POI with LPETG sequence fused to its C-terminal was labeled by fluorophore–pentaglycine conjugation.66 Similarly, pentaglycine on POI's N-terminal can form amide bond with fluorophore–LPETGG oligopeptide under the catalysis of SrtA.67 Recently, Liu group68 reported a sortase-mediated hydrazinolysis of proteins with hydrazine or its derivatives for the production of recombinant protein hydrazides. This strategy provides an convient approach for protein semi-synthesis and an alternative method for C-terminal modification and labeling of proteins.Several other systems, such as N-myristoyl transferase (NMT)–GXXXS tag system and farnesyltransferase (FTase)–CaaX tag system, are also based on the same principle as aforementioned.69
5. Peptide tag containing enzymatic domain for interacting with small molecules or coenzymes
In previous category, the peptides fused to POIs were recognized and linked to probes by endogenous cellular enzymes. As an opposite strategy, enzymes or peptides with enzymatic function are fused to POIs, which can recognize certain kinds of substrates with organic fluorophores. POIs will be labelled when the substrates are linked to the enzyme tag. This class of tags mainly include SNAP-tag, HaloTag, TMP-tag, PYP-tag, BL-tag etc., and have been widely used for protein labelling and modification.The labelling methods of this category own several common advantages. One of the advantages is that we can control the labelling with specific space-time and perform sequential labelling with these tags. For instance, these labelling systems enable pulse-labelling proteins, which allow biologists to track protein populations in exact time scope. The probe containing fluorophores are usually cell-permeable and therefore enable labelling in the whole cell.70 Additionally, since fluorophores in these systems are mostly small molecule chemical ligands, they also possess many advantages over FPs such as higher brightness and more chromatic diversity. Some ligands in these systems also own some advantages like being photoswitchable or sensitive to certain environmental conditions.71 Moreover, most ligands with fluorophore used in these methods own relative good cell permeability, which would contribute to labelling in the whole cell instead of only on the surface. Other advantages include high specificity and fast kinetics, relatively small sizes and so on.
5.1 SNAP-tag and CLIP-tag
Johnsson group72 reported SNAP-tag (human DNA repair protein O6-alkylguanine–DNA alkyltransferase, hAGT) for protein site-specific labelling in 2003. It was the first protein tag to catalyze covalent binding of a fluorophore. hAGT can transfer alkyl group from O6-alkylguanine–DNA to its cysteine residues irreversibly. Because SNAP-tag exploits the low substrate specificity of AGT i.e. AGT can also react with O6-benzylguanine or derivatives of O6-benzylguanosine with substituted benzyl rings. Therefore, in SNAP-tag system, O6-benzylguanine (BG) derivatives are used to label AGT fusion proteins (Fig. 6a). Johnsson group73 have reported a variety of synthetic BG derivatives and exhibited that these AGT substrates can be used for the sequential labelling of AGT fusion proteins.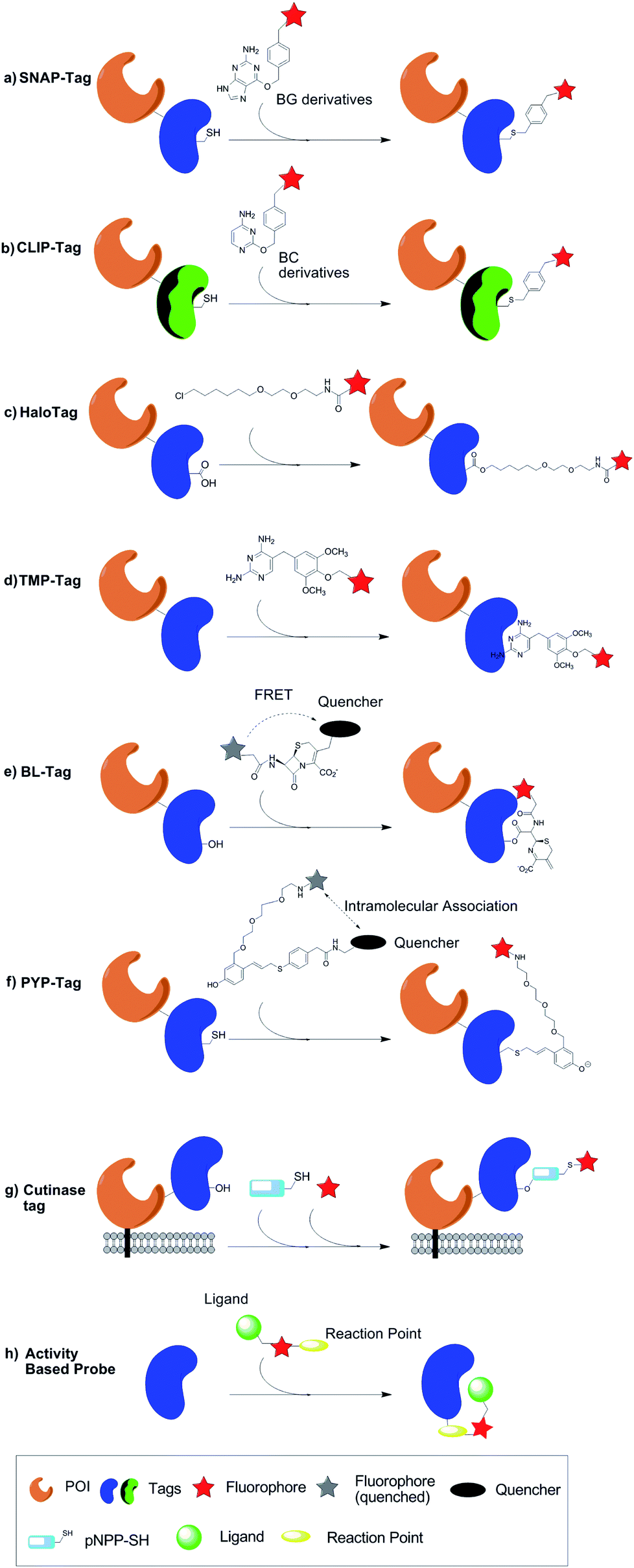 | ||
| Fig. 6 Protein labelling by different peptide tag containing enzymatic domain which can interact with small molecules or coenzymes. | ||
In order to label multiple proteins in a same cell respectively, Johnsson group74 further developed CLIP-tag, which is orthogonal to SNAP-tag. The differences between two tags are the use of different labelling molecules (Fig. 6b). Instead of BG derivatives, CLIP-tag reacts with O2-benzylcytosine (BC) derivatives specifically. Johnsson group also developed a series of BC derivatives that can be used for labelling. These developments promote SNAP-tag and CLIP-tag to be used for FRET measurements or multicolor analysis in a single living cell.
5.2 HaloTag
HaloTag,75 as an enzyme based fusion tag derived from bacterial haloalkane dehalogenase, was developed by researchers from Promega Corporation. Similar to SNAP-tag, HaloTag is another kind of enzyme suicide substrate-based tag. The catalyzing process of wild type haloalkane dehalogenase is as following: aspartate 106 works as nucleophile and displaces halides in the haloalkane, and histidine 272 then reacts with the covalent aspartate-chemical junction, releasing the hydrocarbon chains of the ligand and regenerating the aspartate. The histidine 272 of HaloTag is mutated, which makes the hydrocarbon chain no longer able to get released, and the substrates containing a haloalkane chain are covalently attached to HaloTag76 (Fig. 6c). When the synthetic HaloTag ligand is a chloroalkane linker attached to fluorescent dyes, the tagged protein can be traced by the fluorophore.Derived from an enzyme, HaloTag catalyzes covalent bond formation in a specific and rapid fashion, and the reaction is essentially irreversible.76 Moreover, compared with FPs, HaloTag fluorophores emit more photons and are relatively photostable. These advantages contribute to wide applications of HaloTag. So far 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 human and 15000 mouse genes with HaloTag are commercial available.77 HaloTag-based target-specific azido DCDHFs, a class of photoactivatable push–pull fluorogens have been used for single-molecule super-resolution imaging in live bacterial and fixed mammalian cells.78 Aye group79 recently reported a novel electrophile delivery platform which can be used to deliver biological electrophiles to specific electrophile-responsive proteins. This platform enables precise study of consequences of introducing such chemicals into cells since it can avoid the off-target effect of the excess electrophiles. In this system, HaloTag is exploited to target POIs because of its irreversibility, high specificity and tolerance to sterically demanding groups on the ligands. HaloTag is also a useful approach in single molecule force spectroscopy techniques for anchoring proteins to surfaces due to the high detachment forces and extended lifetime provided by the covalent linkage. Recently the mechanical properties of HaloTag proteins was studied via atomic force microscopy (AFM).80
000 human and 15000 mouse genes with HaloTag are commercial available.77 HaloTag-based target-specific azido DCDHFs, a class of photoactivatable push–pull fluorogens have been used for single-molecule super-resolution imaging in live bacterial and fixed mammalian cells.78 Aye group79 recently reported a novel electrophile delivery platform which can be used to deliver biological electrophiles to specific electrophile-responsive proteins. This platform enables precise study of consequences of introducing such chemicals into cells since it can avoid the off-target effect of the excess electrophiles. In this system, HaloTag is exploited to target POIs because of its irreversibility, high specificity and tolerance to sterically demanding groups on the ligands. HaloTag is also a useful approach in single molecule force spectroscopy techniques for anchoring proteins to surfaces due to the high detachment forces and extended lifetime provided by the covalent linkage. Recently the mechanical properties of HaloTag proteins was studied via atomic force microscopy (AFM).80
5.3 TMP-tag
In 2005, Cornish group81 developed a novel small-molecule inhibitor-based protein labelling system named TMP-tag, in which trimethoprim (TMP) and E. coli dihydrofolate reductase (eDHFR) were used to be a ligand–receptor pair for in vivo imaging. In this system, the target protein is fused with eDHFR through genetic recombination and then labelled by binding to a TMP-fluorophore ligand (Fig. 6d).A great advantage of this labelling system is the relatively low background noise caused by native homologous enzymes. The ligand (TMP) in this system exhibited significantly higher binding affinity to eDHFR than mammalian forms of DHFR (approximately 1000 fold). As a result, the usefulness of TMP–eDHFR does not require a knock-out or modified cell line.82 Based on this system, Cornish group83 developed a covalent variant of TMP-tag in 2009. This covalent bond is formed by a proximity-induced reaction between the eDHFR tag and the TMP–fluorophore. To overcome the low kinetic rate and limited activity for TMP-tag, a second-generation of this covalent labelling system was developed in 2012.84 In this optimized system, a cysteine nucleophile is engineered just outside the TMP-binding pocket of eDHFR, which reacts with added acrylamide–trimethoprim–fluorophore electrophiles. Routine ways to synthesize a variety of probe labels to this system were also presented.84
5.4 BL-tag and PYP-tag
Aforementioned three types of enzyme-tag based protein labelling systems need a washing step to remove free fluorescent probes in case they interfere signal from the target protein. However, the washing step is time-consuming, and the washout of free probes can hardly be completed in 100%.85 To overcome this drawback, researchers have been pursuing fluorogenic labelling systems, in which the labelling probes only show fluorescence after binding to the POIs, and therefore no washing procedure is required.86 Kikuchi group have developed two fluorogenic labelling systems based on different types of enzyme-tags, namely mutant β-lactamase (BL) and photoactive yellow protein (PYP).A series of development90,91 came out following the initial work. In a recent development,89 probes with fast turn-on response have been developed, which broke the major bottlenecks for this approach.
Similar to BL-tag, lower turn-on kinetics of the probe FCTP also prevented the practical application of PYP-tag as a protein labelling system. Recently, Kikuchi group85 designed two new probes based on a novel scaffold to overcome abovementioned shortcoming. In this scaffold, cinnamic acid thioester served as a PYP-tag ligand and nitrobenzene worked as the quencher. This new system significantly speeded up the turn-on process and therefore enabled PYP-tag to be utilized in more complex occasions.
5.5 Cutinase tag
Andrian group93 utilized fungal enzyme cutinase to label proteins on the surface of cell membrane. Cutinase is a serine esterase with a molecular weight of around 22 kDa. p-Nitrophenyl phosphonate (pNPP), a suicide inhibitor of cutinase, reacts with the key catalytic residue Ser120 at the active site of the enzyme, forming a covalent linkage in between. When the pNPP carries a reactive sulfhydryl group at the end of its alkyl chain (pNPP–SH), fluorochromes can be subsequently conjugated to the enzyme. As an example, the group labelled integrin lymphocyte function-associated antigen-1 (LFA-1). They inserted cutinase into the extracellular domain of LFA-1, and showed that the resulting fusion-protein did not affect the normal function of the target protein. They also demonstrated efficiency specificity of the binding between the fluorochrome and the target protein.An important advantage of the strategy is that it allows using quantum dots as fluorochromes, which provide more intense and more stable multiphoton fluorescence.94 Moreover, compared with other enzyme tags, background noise of this strategy is relatively low because of no homologous protein of cutinase in mammals. However, since pNPP–SH cannot permeate the cell membrane, the application of this labelling strategy is limited to extracellular proteins.
Although enzymatic tag mediated protein labelling systems are being developed rapidly and more variants and new ligands are offered every year, some common limitations still exist. First, the size of tag remains large and sometimes can interfere with normal cellular or enzymatic function. It is obvious to note that even the smallest PYP-tag has a size of 14 kDa. In addition, except for BL-tag and PYP-based labelling systems, most tags need a washing step, which is troublesome as described in 5.4. Endogenous enzymes that have similar catalytic specificity might also react with the corresponding ligands, creating high background noise. For example, native AGT in mammalian cells95 may be able to react with SNAP-tag ligands.
It should be mentioned that people have developed specific affinity-based labelling methods for each enzymes. In this strategy, the fluorescence probes are also covalently linked to the substrates of the enzymes. However, the substrate targeting the active site only plays a role in guiding the probe to form covalent bond with an amino acid at another site instead of the active site.96 And this method has been widely used for the identification and characterization of enzymes.97 (Fig. 6g) Based on the similar principle, these enzyme sequences could be fused to POIs as an addition to this category. However, very few of this kind peptide tag has been reported yet. More and more new strategies may also be applied to create enzyme tags with better efficiency and selectivity.
6. Peptide tag labelling with probes by natural or artificial protein–protein interaction
Protein–protein interactions exist widely in biological systems and play center roles in their functions and regulations.98 Considering the selective recognitions between two protein moieties, people have developed several related methods for POIs' orthogonal labelling.6.1 Coiled-coil labelling system
An accessible and convenient way for protein labelling is to make use of the sophisticated protein–protein interaction designed by nature directly. For example, some sequences can form heterodimer coiled-coil rapidly and nontoxically without any metal ion or enzyme. Under this flag, Matsuzaki et al.99 developed Coiled-Coil Labelling method. In this strategy, a cell membrane POI was genetically fused with one of the coiled-coil pairs or an E3 tag with (EIAALEK)3 sequence, while the other component K3 or K4 probe with a sequence of (KIAALKE)n (n = 3 or 4) was labelled with fluorescent dyes. Therefore, the POI was selectively labelled due to the spontaneous formation of heterodimeric coiled-coil structure (Fig. 7). Matsuzaki group have successfully labelled two membrane proteins including human β2-adrenergic receptor and human epidermal growth factor receptor to demonstrate the potential application of this strategy.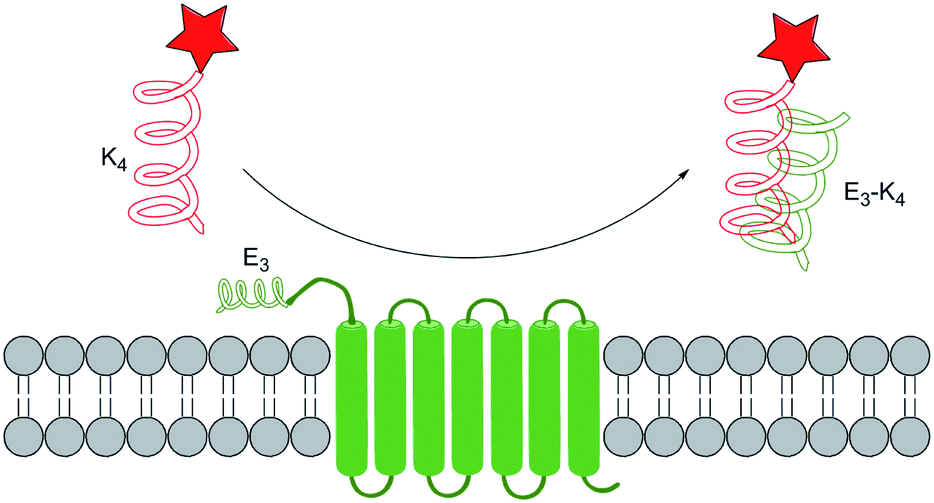 | ||
| Fig. 7 Protein labelling by coiled-coil recognition system. | ||
A wide scope of fluorophores are applicable in this labelling method, and thus we could make choices corresponding according to the need. However, this method owns limitations on the choices of cell lines: cell lines with high negative charges on the membrane surface are not applicable to it because of the high background noise brought by non-specific binding. In order to reduce the positive charges on the probe, Matsuzaki group100 further synthesized and compared TMR–(pSer)n–K4 with a different n vary from 1 to 3. The results showed that n = 1 is the most applicable to different cell lines without heavy aggregation property.
6.2 Leucine zipper peptide labelling system
Leucine zipper sequence has been used for affinity purification of expressed proteins for its high selectivity and high affinity binding.101 Tamamura group102 further applied this specific sequence for protein labelling. They designed tag-probe pairs according to a GCN4 antiparallel coiled-coil trimer crystal structures, and binding of the tag with the peptide leads to a huge fluorescent colorimetric shift. Membrane protein CXCR4 was used as an example to demonstrate that A2-taged protein can fuse to the probe peptide. Compared with the former work with coiled-coil structure or two α-helical leucine zipper, this system have advantages in real-time living cells imaging without the necessity of removing additional probes.6.3 Split native protein labelling systems
Another possible idea for protein labelling is to artificially split a protein into two species that can interact with each other with high affinity. This idea can trace back to the well-known yeast two-hybrid system: the split of GAL4 (ref. 103) can create two parts that can bind to each other. Splitting ubiquitin104 or luciferase105 can also create similar protein pairs. Moreover, yeast three-hybrid system106 can be regarded as an extension of this idea. These techniques have been widely used for protein–protein interaction detection or small molecule screening.Muir and collaborators107 first introduced protein splicing as a efficient tool for protein semisynthesis. Intein is a protein-splicing enzyme which contains all the catalytic activity by itself.108 After the formation of C-extein intein thioester, it can undergo thiol exchange reaction with any cysteine containing moiety. Several examples have been reported to demonstrate this as an applicable protein labelling method with biotin and fluorophores.109
Besides abundant contiguous intein, split intein is a rare group of intein whose C-terminal and N-terminal can spontaneously assemble.110 The specific binding between the two parts of split intein111 triggers the protein splicing process which results in the cleavage of intein and the formation of a POI linked with native amide bond (Fig. 8). Split intein strategy has been used in several labelling methods such as introducing isotope labelling groups into proteins for the NMR study,112 fluorophores for FRET research113 and so on. Although efforts have been made to develop other split protein pairs, to our best knowledge only split intein has been used for protein labelling research till now.
 | ||
| Fig. 8 Protein labelling by split intein systems. | ||
7. Conclusion
Proteins are of primary importance in almost all kinds of life activities. Therefore, to study the roles of proteins under physiological condition with exact spatiotemporal control is always tantalizing for scientists. Along with the development of molecular biology, more and more in vivo labelling methods have been developed based on gene engineering techniques. The most common form is a peptide (or protein) sequence additionally attached to POIs, which can be directly or indirectly labelled at certain sites.114 These processes often have convincing efficiency and bio-orthogonality, guaranteed by specificity of enzyme recognition or selectivity of covalent bond formation. There are already many applications of these labelling strategies.115 This review divided them into five categories and introduced each of them based on different principles.We are looking forward to see more and more convenient, robust, and efficient methods to label proteins in the future. Combined with the total and semi-chemical protein synthesis methods to study proteins under in vitro conditions,116 these chemistry-enabled technologies are expected to greatly impact the future studies on structures and functions of proteins.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21102083 and 21372058 to Y. M. L.).Notes and references
- L. W. Miller and V. W. Cornish, Curr. Opin. Chem. Biol., 2005, 9, 56 CrossRef CAS PubMed.
- H. Sahoo, RSC Adv., 2012, 2, 7017 RSC.
- R. Wombacher and V. W. Cornish, J. Biophotonics, 2011, 4, 391 CrossRef CAS PubMed.
- G. Crivat and J. W. Taraska, Trends Biotechnol., 2012, 30, 8 CrossRef CAS PubMed.
- M. Rashidian, J. K. Dozier and M. D. Distefano, Bioconjugate Chem., 2013, 24, 1277 CrossRef CAS PubMed.
- C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith and P. G. Schultz, Science, 1989, 244, 182 CAS.
- (a) J. Xie and P. G. Schultz, Nat. Rev. Mol. Cell Biol., 2006, 7, 775 CrossRef CAS PubMed; (b) L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498 CrossRef CAS PubMed; (c) J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. Zhang and P. G. Schultz, Science, 2003, 301, 964 CrossRef CAS PubMed; (d) Y. M. Li, M. Y. Yang, Y. C. Huang, X. D. Song, L. Liu and P. R. Chen, Chem. Sci., 2012, 3, 2766 RSC; (e) Y. M. Li, M. Y. Yang, Y. C. Huang, Y. T. Li, P. R. Chen and L. Liu, ACS Chem. Biol., 2012, 7, 1015 CrossRef CAS PubMed; (f) Y. M. Li, M. Pan, Y. T. Li, Y. C. Huang and Q. X. Guo, Org. Biomol. Chem., 2013, 11, 2624 RSC; (g) D. Mendel, V. W. Cornish and P. G. Schultz, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 435 CrossRef CAS PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509 CrossRef CAS PubMed.
- O. Shimomura, F. H. Johnson and Y. Saiga, J. Cell. Comp. Physiol., 1962, 59, 223 CrossRef CAS.
- D. C. Prasher, V. K. Eckenrode, W. W. Ward, F. G. Prendergast and M. J. Cormier, Gene, 1992, 111, 229 CrossRef CAS.
- M. Chalfie, Y. Tu, G. Euskirchen, W. Ward and D. Prasher, Science, 1994, 263, 802 CAS.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217 CrossRef CAS PubMed.
- J. Lippincott-Schwartz and G. H. Patterson, Science, 2003, 300, 87–91 CrossRef CAS PubMed.
- J.-D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79 CrossRef PubMed.
- S. Cabantous, Y. Rogers, T. C. Terwilliger and G. S. Waldo, PLoS One, 2008, 3, e2387 Search PubMed.
- A. R. Nager, T. A. Baker and R. T. Sauer, J. Mol. Biol., 2011, 413, 4 CrossRef CAS PubMed.
- R. Heim, D. C. Prasher and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12501 CrossRef CAS.
- M. Ormö, a. B. Cubitt, K. Kallio, L. a. Gross, R. Y. Tsien and S. J. Remington, Science, 1996, 273, 1392 Search PubMed.
- D. M. Shcherbakova, O. M. Subach and V. V. Verkhusha, Angew. Chem., Int. Ed. Engl., 2012, 51, 10724 CrossRef CAS PubMed.
- A. S. Mishin, F. V. Subach, I. V. Yampolsky, W. King, K. A. Lukyanov and V. V. Verkhusha, Biochemistry, 2008, 47, 4666 CrossRef CAS PubMed.
- M. V Matz, A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov and S. A. Lukyanov, Nat. Biotechnol., 1999, 17, 969 CrossRef PubMed.
- R. E. Campbell, O. Tour, A. E. Palmer, P. a. Steinbach, G. S. Baird, D. a. Zacharias and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7877 CrossRef CAS PubMed.
- L. a. Gross, G. S. Baird, R. C. Hoffman, K. K. Baldridge and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11990 CrossRef CAS PubMed.
- P. Corish and C. Tyler-Smith, Protein Eng., 1999, 12, 1035 CrossRef CAS PubMed.
- P. Dedecker, F. C. De Schryver and J. Hofkens, J. Am. Chem. Soc., 2013, 135, 2387 CrossRef CAS PubMed.
- O. Dyachok, Y. Isakov, J. Sågetorp and A. Tengholm, Nature, 2006, 439, 349 CrossRef CAS PubMed.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269 CrossRef CAS.
- B. Albert Griffin, S. R. Adams, J. Jones and R. Y. Tsien, Methods Enzymol., 2000, 327, 565 Search PubMed.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063 CrossRef CAS PubMed.
- A. Y. Ting, K. H. Kain, R. L. Klemke and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 15003 CrossRef CAS PubMed.
- T. A. Cornell, J. Fu, S. H. Newland and B. P. Orner, J. Am. Chem. Soc., 2013, 135, 16618 CrossRef CAS PubMed.
- T. L. Halo, J. Appelbaum, E. M. Hobert, D. M. Balkin and A. Schepartz, J. Am. Chem. Soc., 2008, 131, 438 CrossRef PubMed.
- M. Hedhammar, T. Gräslund and S. Hober, Chem. Eng. Technol., 2005, 28, 1315 CrossRef CAS.
- E. Hochuli, H. Dobeli and A. Schacher, J. Chromatogr., A, 1987, 411, 177 CrossRef CAS.
- E. G. Guignet, R. Hovius and H. Vogel, Nat. Biotechnol., 2004, 22, 440 CrossRef CAS PubMed.
- C. R. Goldsmith, J. Jaworski, M. Sheng and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 418 CrossRef CAS PubMed.
- A. Ojida, K. Honda, D. Shinmi, S. Kiyonaka, Y. Mori and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 10452 CrossRef CAS PubMed.
- K. Honda, E. Nakata, A. Ojida and I. Hamachi, Chem. Commun., 2006, 4024 RSC.
- H. Nonaka, S.-H. Fujishima, S.-H. Uchinomiya, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 9301 CrossRef CAS PubMed.
- A. Ojida, S.-H. Fujishima, K. Honda, H. Nonaka, S.-H. Uchinomiya and I. Hamachi, Chem. – Asian J., 2010, 5, 877 CrossRef CAS PubMed.
- S.-H. Fujishima, H. Nonaka, S.-H. Uchinomiya, Y. A. Kawase, A. Ojida and I. Hamachi, Chem. Commun., 2012, 48, 594 RSC.
- C. T. Hauser and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3693 CrossRef CAS PubMed.
- J. M. Antos, J. M. McFarland, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 6301 CrossRef CAS PubMed.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256 CrossRef CAS PubMed.
- D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC.
- M. Fernández-Suárez, H. Baruah, L. Martínez-Hernández, K. T. Xie, J. M. Baskin, C. R. Bertozzi and A. Y. Ting, Nat. Biotechnol., 2007, 25, 1483 CrossRef PubMed.
- C. Uttamapinant, K. A. White, H. Baruah, S. Thompson, M. Fernández-Suárez, S. Puthenveetil and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10914 CrossRef CAS PubMed.
- X. Jin, C. Uttamapinant and A. Y. Ting, ChemBioChem, 2011, 12, 65 CrossRef CAS PubMed.
- D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792 CrossRef CAS PubMed.
- J. Z. Yao, C. Uttamapinant, A. Poloukhtine, J. M. Baskin, J. A. Codelli, E. M. Sletten, C. R. Bertozzi, V. V. Popik and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 3720–3728 CrossRef CAS PubMed.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Angew. Chem., Int. Ed. Engl., 2012, 51, 5852–5856 CrossRef CAS PubMed.
- J. D. Cohen, P. Zou and A. Y. Ting, ChemBioChem, 2012, 13, 888–894 CrossRef CAS PubMed.
- S. Puthenveetil, D. S. Liu, K. A. White, S. Thompson and A. Y. Ting, J. Am. Chem. Soc., 2009, 131, 16430 CrossRef CAS PubMed.
- D. S. Liu, W. S. Phipps, K. H. Loh, M. Howarth and A. Y. Ting, ACS Nano, 2012, 6, 11080 CAS.
- H. Baruah, S. Puthenveetil, Y. A. Choi, S. Shah and A. Y. Ting, Angew. Chem., Int. Ed. Engl., 2008, 47, 7018 CrossRef CAS PubMed.
- Y. Sugimura, M. Hosono, F. Wada, T. Yoshimura, M. Maki and K. Hitomi, J. Biol. Chem., 2006, 281, 17699 CrossRef CAS PubMed.
- C. W. Lin and A. Y. Ting, J. Am. Chem. Soc., 2006, 128, 4542 CrossRef CAS PubMed.
- M. Howarth, K. Takao, Y. Hayashi and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7583 CrossRef CAS PubMed.
- M. Howarth and A. Y. Ting, Nat. Protoc., 2008, 3, 534 CrossRef CAS PubMed.
- P. Zou and A. Y. Ting, ACS Chem. Biol., 2011, 6, 308 CrossRef CAS PubMed.
- J. D. Martell, T. J. Deerinck, Y. Sancak, T. L. Poulos, V. K. Mootha, G. E. Sosinsky, M. H. Ellisman and A. Y. Ting, Nat. Biotechnol., 2012, 30, 1143 CrossRef CAS PubMed.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929 CrossRef PubMed.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321 CrossRef CAS PubMed.
- D. Rabuka, J. S. Rush, G. W. de Hart, P. Wu and C. R. Bertozzi, Nat. Protoc., 2012, 7, 1052 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670 CrossRef CAS PubMed.
- M. W. Popp, J. M. Antos, G. M. Grotenbreg, E. Spooner and H. L. Ploegh, Nat. Chem. Biol., 2007, 3, 707 CrossRef CAS PubMed.
- T. Yamamoto and T. Nagamune, Chem. Commun., 2009, 1022 RSC.
- Y. M. Li, Y. T. Li, M. Pan, X.-Q. Kong, Y.-C. Huang, Z.-Y. Hong and L. Liu, Angew. Chem., Int. Ed. DOI:10.1002/anie.201310010.
- M. Sunbul and J. Yin, Org. Biomol. Chem., 2009, 7, 3361 CAS.
- R. Wombacher and V. W. Cornish, J. Biophotonics, 2011, 4, 391 CrossRef CAS PubMed.
- G. Crivat and J. W. Taraska, Trends Biotechnol., 2012, 30, 8 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86 CrossRef CAS PubMed.
- A. Keppler, H. Pick, C. Arrivoli, H. Vogel and K. Johnsson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9955 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128 CrossRef CAS PubMed.
- G. V. Los and K. Wood, Methods Mol. Biol., 2007, 356, 195 CAS.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373 CrossRef CAS PubMed.
- For example: http://www.genecopoeia.com/tech/halo-tag/, accessed November 7, 2013.
- H. D. Lee, S. J. Lord, S. Iwanaga, K. Zhan, H. Xie, J. C. Williams, H. Wang, G. R. Bowman, E. D. Goley, L. Shapiro, R. J. Twieg, J. Rao and W. E. Moerner, J. Am. Chem. Soc., 2010, 132, 15099 CrossRef CAS PubMed.
- X. Fang, Y. Fu, M. J. C. Long, J. a. Haegele, E. J. Ge, S. Parvez and Y. Aye, J. Am. Chem. Soc., 2013, 135, 14496 CrossRef CAS PubMed.
- I. Popa, R. Berkovich, J. Alegre-Cebollada, C. L. Badilla, J. A. Rivas-Pardo, Y. Taniguchi, M. Kawakami and J. M. Fernandez, J. Am. Chem. Soc., 2013, 135, 12762 CrossRef CAS PubMed.
- L. W. Miller, Y. Cai, M. P. Sheetz and V. W. Cornish, Nat. Methods, 2005, 2, 255 CrossRef CAS PubMed.
- N. T. Calloway, M. Choob, A. Sanz, M. P. Sheetz, L. W. Miller and V. W. Cornish, ChemBioChem, 2007, 8, 767 CrossRef CAS PubMed.
- S. S. Gallagher, J. E. Sable, M. P. Sheetz and V. W. Cornish, ACS Chem. Biol., 2009, 4, 547 CrossRef CAS PubMed.
- Z. Chen, C. Jing, S. S. Gallagher, M. P. Sheetz and V. W. Cornish, J. Am. Chem. Soc., 2012, 134, 13692 CrossRef CAS PubMed.
- Y. Hori, K. Nakaki, M. Sato, S. Mizukami and K. Kikuchi, Angew. Chem., Int. Ed. Engl., 2012, 51, 5611 CrossRef CAS PubMed.
- S. Mizukami, Y. Hori and K. Kikuchi, Acc. Chem. Res., 2013 DOI:10.1021/ar400135f.
- S. Mizukami, S. Watanabe, Y. Hori and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 5016 CrossRef CAS PubMed.
- H. Adachi, T. Ohta and H. Matsuzawa, J. Biol. Chem., 1991, 266, 3186 CAS.
- S. Mizukami, S. Watanabe, Y. Akimoto and K. Kikuchi, J. Am. Chem. Soc., 2012, 134, 1623 CrossRef CAS PubMed.
- S. Watanabe, S. Mizukami, Y. Hori and K. Kikuchi, Bioconjugate Chem., 2010, 21, 2320 CrossRef CAS PubMed.
- S. Watanabe, S. Mizukami, Y. Akimoto, Y. Hori and K. Kikuchi, Chemistry, 2011, 17, 8342 CrossRef CAS PubMed.
- Y. Hori, H. Ueno, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 16610 CrossRef CAS PubMed.
- R. Bonasio, C. V. Carman, E. Kim, P. T. Sage, K. R. Love, T. R. Mempel, T. a. Springer and U. H. von Andrian, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14753 CrossRef CAS PubMed.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86 CrossRef CAS PubMed.
- L. Wofsy, H. Metzger and S. Singer, Biochemistry, 1962, 1, 1031 CrossRef CAS.
- Y. Takaoka, A. Ojida and I. Hamachi, Angew. Chem., Int. Ed. Engl., 2013, 52, 4088 CrossRef CAS PubMed.
- H.-K. Cui, J. Qing, Y. Guo, Y.-J. Wang, L.-J. Cui, T.-H. He, L. Zhang and L. Liu, Bioorg. Med. Chem., 2013, 21, 3547 CrossRef CAS PubMed.
- Y. Yano, A. Yano, S. Oishi, Y. Sugimoto, G. Tsujimoto, N. Fujii and K. Matsuzaki, ACS Chem. Biol., 2008, 3, 341 CrossRef CAS PubMed.
- S. Ono, Y. Yano and K. Matsuzaki, Biopolymers, 2012, 98, 234 CrossRef CAS PubMed.
- (a) B. Tripet, L. Yu, D. L. Bautista, W. Y. Wong, R. T. Irvin and R. S. Hodges, Protein Eng., 1996, 9, 1029 CrossRef CAS PubMed; (b) K. Zhang, M. R. Diehl and D. A. Tirrell, J. Am. Chem. Soc., 2005, 127, 10136 CrossRef CAS PubMed; (c) S. Yuzawa, T. Mizuno and T. Tanaka, Chem. – Eur. J., 2006, 12, 7345 CrossRef CAS PubMed.
- H. Tsutsumi, W. Nomura, S. Abe, T. Mino, A. Masuda, N. Ohashi, T. Tanaka, K. Ohba, N. Yamamoto and K. Akiyoshi, Angew. Chem., Int. Ed. Engl., 2009, 121, 9328 CrossRef.
- S. Fields and O. Song, Nature, 1989, 340, 245 CrossRef CAS PubMed.
- N. Johnsson and A. Varshavsky, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10340 CrossRef CAS.
- T. Ozawa, A. Kaihara, M. Sato, K. Tachihara and Y. Umezawa, Anal. Chem., 2001, 73, 2516 CrossRef CAS.
- S. S. Gallagher, L. W. Miller and V. W. Cornish, Anal. Biochem., 2007, 363, 160 CrossRef CAS PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705 CrossRef CAS.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249 CrossRef CAS PubMed.
- S. Chattopadhaya, L. P. Tan and S. Q. Yao, Nat. Protoc., 2006, 1, 2386 CrossRef CAS PubMed.
- N. H. Shah, E. Eryilmaz, D. Cowburn and T. W. Muir, J. Am. Chem. Soc., 2013, 135, 18673 CrossRef CAS PubMed.
- N. H. Shah and T. W. Muir, Isr. J. Chem., 2011, 51, 854 CrossRef CAS PubMed.
- S. Züger and H. Iwai, Nat. Biotechnol., 2005, 23, 736 CrossRef PubMed.
- J.-Y. Yang and W. Y. Yang, J. Am. Chem. Soc., 2009, 131, 11644 CrossRef CAS PubMed.
- D. Jung, K. Min, J. Jung, W. Jang and Y. Kwon, Mol. BioSyst., 2013, 9, 862 RSC.
- C. Jing and V. W. Cornish, Acc. Chem. Res., 2011, 44, 784 CrossRef CAS PubMed.
- (a) P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776 CAS; (b) S. B. Kent, Chem. Soc. Rev., 2009, 38, 338 RSC; (c) S. B. Kent, Angew. Chem., Int. Ed. Engl., 2013, 52, 11988 CrossRef CAS PubMed; (d) J. S. Zheng, S. Tang, Y. C. Huang and L. Liu, Acc. Chem. Res., 2013, 46, 2475 CrossRef CAS PubMed; (e) P. Wang, S. Dong, J. A. Brailsford, K. Iyer, S. D. Townsend, Q. Zhang, R. C. Hendrickson, J. Shieh, M. A. Moore and S. J. Danishefsky, Angew. Chem., Int. Ed. Engl., 2012, 124, 11744 CrossRef; (f) K. Mandal, M. Uppalapati, D. Ault-Riche, J. Kenney, J. Lowitz, S. S. Sidhu and S. B. Kent, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14779 CrossRef CAS PubMed; (g) G. M. Fang, J. X. Wang and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 10347 CrossRef CAS PubMed; (h) P. Wang, B. Aussedat, Y. Vohra and S. J. Danishefsky, Angew. Chem., Int. Ed. Engl., 2012, 124, 11739 CrossRef; (i) M. Vila-Perello, Z. Liu, N. H. Shah, J. A. Willis, J. Idoyaga and T. W. Muir, J. Am. Chem. Soc., 2013, 135, 286–292 CrossRef CAS PubMed; (j) N. H. Shah, G. P. Dann, M. Vila-Perelló, Z. Liu and T. W. Muir, J. Am. Chem. Soc., 2012, 134, 11338 CrossRef CAS PubMed; (k) J. S. Zheng, H. N. Chang, F. L. Wang and L. Liu, J. Am. Chem. Soc., 2011, 133, 11080 CrossRef CAS PubMed; (l) S. Dong, S. Shang, J. Li, Z. Tan, T. Dean, A. Maeda, T. J. Gardella and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 15122 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |