
Effect of solvent on the uncatalyzed synthesis of aminosilane-functionalized graphene†
Abstract
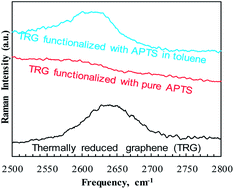
Uncatalyzed functionalization of thermally reduced graphene (TRG) with 3-aminopropyltriethoxy silane (APTS) is reported and the effect of the solvent on selective functionalization is discussed. The chemical, morphological and thermal properties of the functionalized TRG (f-TRG) have been studied using FTIR, XPS, EELS, Raman spectroscopy, TEM, and TGA. Our results indicate that the use of organic solvent during the silylation reaction not only increases grafting yield from 7 to 8 atomic% of Si attachment but also directs APTS groups to the edges of TRG as revealed using energy filtered TEM elemental mapping. A reaction mechanism based on attachment of the silane groups on the TRG surface through residual, surface bound phenolic and carbonyl groups is proposed and discussed. The present approach provides an economical route for mass production of APTS-f-TRG and sheds light on the role of organic solvents in silane functionalization of graphene.
 Please wait while we load your content...
Please wait while we load your content...

