 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the role of fluoro-substituted nucleosides in DNA radiosensitization for tumor radiation therapy
J. Kopyraa,
A. Kellerb and
I. Bald*cd
aDepartment of Chemistry, Siedlce University, 3 Maja 54, 08-110 Siedlce, Poland
bInstitute of Ion Beam Physics and Materials Research, Helmholtz-Zentrum Dresden-Rossendorf, Dresden, Germany
cInstitute of Chemistry – Physical Chemistry, University of Potsdam, Germany. E-mail: ilko.bald@uni-potsdam.de
dBAM Federal Institute of Materials Research and Testing, Berlin, Germany
First published on 6th January 2014
Abstract
Gemcitabine (2′,2′-difluorocytidine) is a well-known radiosensitizer routinely applied in concomitant chemoradiotherapy. During irradiation of biological media with high-energy radiation secondary low-energy (<10 eV) electrons are produced that can directly induce chemical bond breakage in DNA by dissociative electron attachment (DEA). Here, we investigate and compare DEA to the three molecules 2′-deoxycytidine, 2′-deoxy-5-fluorocytidine, and gemcitabine. Fluorination at specific molecular sites, i.e., nucleobase or sugar moiety, is found to control electron attachment and subsequent dissociation pathways. The presence of two fluorine atoms at the sugar ring results in more efficient electron attachment to the sugar moiety and subsequent bond cleavage. For the formation of the dehydrogenated nucleobase anion, we obtain an enhancement factor of 2.8 upon fluorination of the sugar, whereas the enhancement factor is 5.5 when the nucleobase is fluorinated. The observed fragmentation reactions suggest enhanced DNA strand breakage induced by secondary electrons when gemcitabine is incorporated into DNA.
Introduction
The majority of patients diagnosed with cancer receive a radiation therapy treatment to reduce or remove the tumor tissue. 5-fluorouracil, cisplatin (cis-diamminedichloroplatinum(II)) and 2′,2′-difluorocytidine (gemcitabine, Gem) belong to the most widely applied therapeutics in combined chemoradiotherapy, which are administered concomitantly with the high-energy radiation treatment to increase the effect of the radiation.1 The biological effect of these radiosensitizers is the inhibition of DNA replication by different mechanisms.2,3 On the other hand, it was shown that cisplatin and halogenated uracils also act on a physicochemical level by direct interaction with low-energy secondary electrons4–7 which are produced in large quantities along the track of the primary high-energy radiation.8,9 Low-energy electrons (LEE) induce DNA strand breaks very efficiently by dissociative electron attachment (DEA) already at very low energies (0–15 eV).10 The cross section for electron-induced single strand breaks (SSBs) has a global maximum at 1 eV,11 whereas the cross section for double strand breaks (DSBs) has a distinct maximum at 10 eV.10,12,13The LEE induced DNA strand breakage is directly enhanced by cisplatin and its derivatives.14 Furthermore, gas-phase DEA measurements have shown that cisplatin reacts with LEEs close to zero eV resulting in a release of both chlorine atoms after electron attachment thereby presumably facilitating the DNA binding.15 After activation, cisplatin binds to the N7 sites of neighboring guanine (G) bases resulting in intra- and interstrand cross links and subsequent cell damage.16
Halogenated uracils are incorporated into the DNA and lead to radiosensitization due to an enhancement of the electron attachment and dissociation cross sections.17 5-Bromouracil and 5-iodouracil appear to be particularly effective, since Br− and I− are formed by electron attachment at zero eV with cross sections of 4 × 10−14 cm2 and 9 × 10−14 cm2, respectively.7,18,19
Gem is a widely applied therapeutic due to its effectiveness towards a broad range of tumors. Although its biological effect as an inhibitor of DNA synthesis and repair is well-studied,2 a radiosensitization due to direct interaction of Gem with secondary electrons has never been considered. Gem is a derivative of 2′-deoxycytidine (dCyt), in which the two hydrogen atoms at C2′ are replaced by two F atoms (Fig. 1). The presence of F atoms usually increases the electron attachment cross sections of the respective molecule considerably. Therefore, in Gem we expect a specific radiosensitization of the sugar unit, as opposed to halogenated purines and pyrimidines, in which the nucleobase is sensitized.17
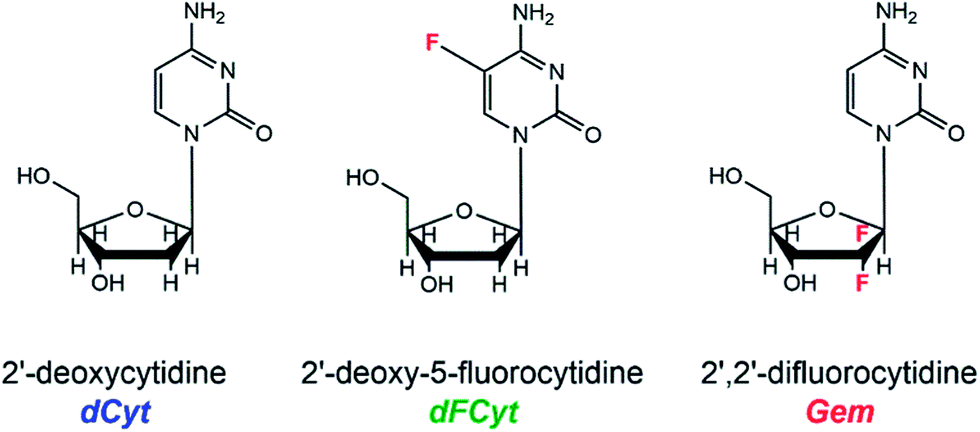 | ||
| Fig. 1 Molecular structures of 2′-deoxycytidine (dCyt), 2′-deoxy-5-fluorocytidine (dFCyt) and 2′,2′-difluorocytidine, which is referred to as gemcitabine (Gem). | ||
In a number of experimental and theoretical DEA studies on small DNA model compounds the question was discussed, whether strand breakage is due to initial electron attachment to the DNA nucleobases,20 or directly to the DNA backbone.21 For strand breakage to happen after LEE attack at the nucleobase the excess charge must be transferred to the sugar-phosphate backbone.22 Despite the large research effort, it could not yet be clarified satisfyingly, which pathway dominates. The most conclusive answer so far is that both mechanisms are operative at different energies as was demonstrated in a recent DEA study on the nucleotide 2′-deoxycytidine-5-monophosphate.23
Here, we probe the competing pathways of electron attachment to either the nucleobase subunit or the sugar unit by studying DEA to the nucleoside dCyt and compare the DEA spectra with the ones obtained from 2′-deoxy-5-fluorocytidine (dFCyt) and Gem (Fig. 1). Our comparative study demonstrates how the reaction pathways can be shifted by fluorination, thus opening up the possibility to control the reaction dynamics by rational chemical design. Furthermore, we demonstrate for the first time an enhanced reactivity of Gem towards LEEs which might contribute to its radiosensitizing properties. The improved understanding of the physicochemical mechanisms of radiosensitization will support a rational and thus efficient optimization of therapeutically used radiosensitizers.
Experimental
DEA to dCyt, dFCyt and Gem was measured using a crossed electron/molecular beam setup housed in a high-vacuum chamber with a base pressure of 10−9 mbar.24 A trochoidal electron monochromator provided an electron beam with an electron energy resolution of 250–300 meV at a current of 10–15 nA. The electron energy scale was calibrated by means of the near 0 eV resonance of the electron scavenger SF6 that was introduced into the vacuum chamber prior and subsequent to a series of DEA measurements. dCyt, dFCyt and Gem were transported into the gas phase by thermal evaporation at 130–190 °C and care was taken to avoid thermal decomposition of the samples by checking 1H NMR spectra of the samples in the container prior and subsequent to the measurements. The anions formed in the reaction volume were mass analyzed with a quadrupole mass spectrometer (QMS) and the intensity of individual fragment anions was recorded as a function of the electron energy in the range of 0–12 eV. All spectra of dCyt and dFCyt have been recorded in a 4 s time mode while in the case of Gem in 2 s time mode. Enhancement factors (EF) for spectra in Fig. 2 have been obtained using integral intensities of the near 0 eV and 1.5 eV resonances and by normalizing the signal of the 1.5 eV resonance of Gem and the 0 eV resonance of dFCyt to the respective resonance of dCyt: EF0 eV,Gem = (I0 eV,Gem/I0 eV,dCyt)(I1.5 eV,dCyt/I1.5 eV,Gem); EF1.5 eV,dFCyt = (I1.5 eV,dFCyt/I1.5 eV,dCyt)(I0 eV,dCyt/I0 eV,dFCyt). The samples of dCyt and Gem (delivered as gemcitabine hydrochloride) were obtained from Sigma-Aldrich (with a stated purity ≥99% and ≥98%, respectively) and dFCyt from TCI (with a stated purity >98%) and used as delivered.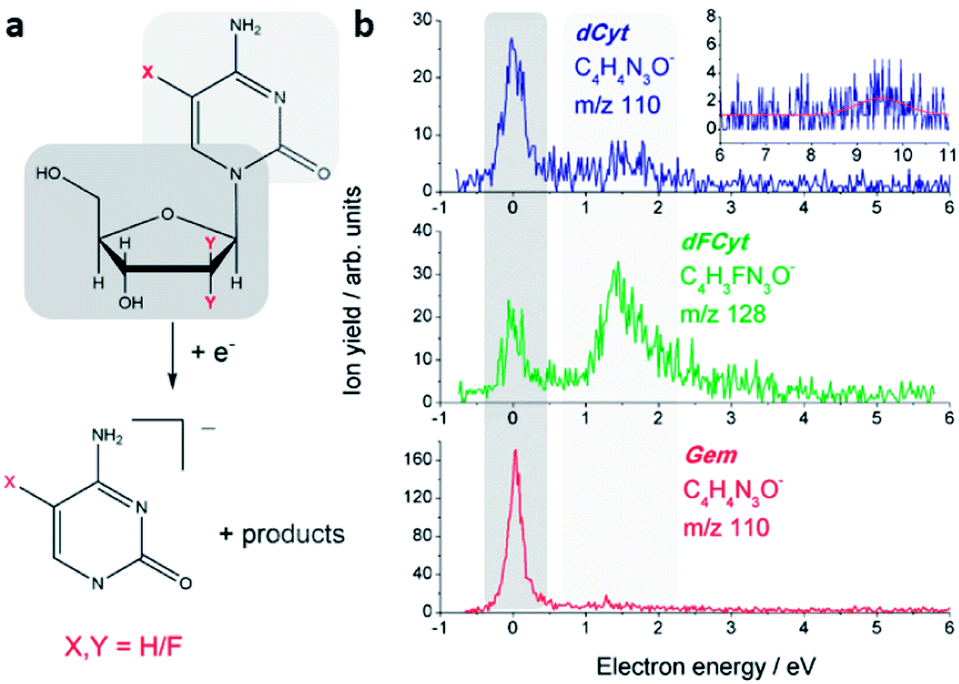 | ||
| Fig. 2 (a) Reaction scheme of the electron-induced N-glycosidic bond cleavage in the three molecules dCyt (X, Y = H), Gem (X = H, Y = F) and dFCyt (X = F, Y = H) with the extra charge remaining on the nucleobase. (b) Corresponding ion yield curves of the respective nucleobase anions that are formed by N-glycosidic bond cleavage. The signal close to zero eV is assigned to initial electron attachment to the sugar unit (gray background), whereas the signal around 1.5 eV is due to electron attachment to the nucleobase (light gray background). | ||
Results and discussion
The initial step in DEA is the formation of a transient negative ion (TNI), which may decay by unimolecular dissociation into a negatively charged species and neutral counterparts. DEA to the three investigated compounds results in a multitude of fragment anions, but no parent anions have been observed indicating that their lifetimes with respect to dissociation are too short to be detected by the QMS. In general, all heavier fragment anions (with more than three atoms) are formed from resonances at very low electron energies, i.e. below 3 eV. In this energy regime the TNIs can be assigned as shape resonances, i.e. the transient anions are generated by electron localization in one of the virtual molecular orbitals (MOs) of the parent molecule in its electronic ground state. The observed fragmentation reactions are accompanied by a cleavage of the N-glycosidic bond, and subsequent to the fragmentation reaction the extra charge is either located on the nucleobase site, or on the sugar moiety.From dCyt a fragment ion is observed at m/z 110, which corresponds to the closed shell dehydrogenated cytosine (C, C4H5N3O) anion [C–H]− (Fig. 2a). The ion yield curve in Fig. 2b shows that it is formed through a narrow resonance close to 0 eV, and with lower intensity at 1.5 eV. Similar signals close to 0 eV have been previously observed in electron attachment to the sugars 2-deoxy-D-ribose,25 D-ribose26 and D-fructose,27 the nucleotide 2′-deoxycytidine-5-monophosphate,23 and the DNA backbone surrogate D-ribose-5-phosphate.21 The near 0 eV signals are generally assigned to electron attachment to the sugar unit by initial formation of a dipole-bound doorway state.28 In this case the electron is initially trapped by the high dipole moment of the molecule, and the diffuse dipole bound state may result in dissociation if it couples to a valence bound state. In contrast, the resonance around 1.5 eV is most likely located at the cytosine unit and corresponds to the π* shape resonance observed from isolated cytosine at the same energy.29 Previous measurements on nucleoside analogues have shown that also in more complex systems the resonances of the isolated building blocks prevail.30 Another weak signal from m/z 110 is observed at 9.5 eV (see inset of Fig. 2), which is most likely due to a core excited resonance located at the cytosine subunit.
A corresponding anion was also observed from dFCyt and Gem. In the case of dFCyt the fragment anion is observed at m/z 128 since it additionally carries a fluorine atom. The dehydrogenated nucleobase anion was observed from all three compounds at the same electron energies, however, the intensity ratios of the two resonances are remarkably different (Fig. 2b). In dFCyt the intensity of the 1.5 eV resonance is considerably higher than in dCyt, and the count rates suggest that it is indeed an increase of the fragmentation rate from the 1.5 eV resonance rather than a simple change of the intensity ratio. The [C–H]− anion formed from Gem is formed with high intensity close to zero eV whereas the 1.5 eV resonance appears to be unchanged compared to dCyt. Comparing the three different compounds the energetic positions of the π* and the zero eV resonance does not change upon fluorination of the corresponding subunit, but the DEA cross section increases considerably. A direct comparison of the signal intensities of different compounds is difficult and might result in large errors. Nevertheless, the enhancement of DEA cross section for the formation of the dehydrogenated nucleobase anion upon fluorination can be quantified under the assumption that only the resonance associated with the fluorinated subunit is modified, whereas the DEA cross section for the other resonance remains constant. Under this assumption we obtain an enhancement factor of EF0 eV,Gem = 2.8 for the near 0 eV resonance in Gem, and EF1.5 eV,dFCyt = 5.5 for the 1.5 eV resonance in dFCyt.
In the case of the close to 0 eV resonance in Fig. 2 it should be noted that there is an efficient transfer of the excess charge from the sugar unit to the cytosine unit. That is, the fluorination at the sugar moiety in Gem leads to 2.8 times more effective electron attachment to the sugar while the charge gets localized on the cytosine moiety after dissociation of the N-glycosidic bond. Based on previous DEA investigations of phosphate containing compounds it is likely that in the presence of a phosphate group an electron transfer from the sugar unit to the phosphate group also occurs resulting in phosphoester cleavage representing a direct DNA strand break.21,23 Such a reaction is driven by the high electron affinity of the phosphate group.
The complementary anion of the dehydrogenated base is the deoxyribose anion (dR−, here it must be noted that in the deoxyribose anion formed from free deoxyribose there is one more hydroxyl group present, which is replaced by the nucleobase in nucleosides), which is not stable enough to be observable in the mass spectrometer.25 Nevertheless, as shown in Fig. 3, from Gem we observed a corresponding anion including the two fluorine atoms indicating that the fragment anion is stabilized by fluorine:
| Gem + e− (0.5 eV) → C5H7F2O3−(m/z 153) + [C–H] |
 | ||
| Fig. 3 Ion yield curves that are due to N-glycosidic bond cleavage with the charge remaining on the sugar unit. Only in the case of Gem an intact sugar anion is observed with small signal intensity (at m/z 153). Nevertheless, the ion with highest signal intensity is due to additional abstraction of neutral C2H2O2 (m/z 94). In the case of dCyt and dFCyt the sugar fragments further dissociate by abstraction of a neutral water molecule. | ||
This is plausible since fluorine generally increases the electron affinity of the fragments. The fluorinated sugar anion is observed with very low intensity at 0.5 eV. From dCyt and dFCyt a signal close to zero eV was detected that is due to additional loss of water and a hydrogen atom (Fig. 3):
| dCyt + e− (≈0 eV) → C5H6O2−(m/z 98) + C + H2O |
| dFCyt + e− (≈0 eV) → C5H6O2−(m/z 98) + FC + H2O |
In the case of dFCyt the signal is almost three times as intense as that of dCyt indicating an increase of the electron attachment cross section upon fluorination at the cytosine. A corresponding fragment was not observed from Gem, but instead a fragment anion at m/z 94 was observed with very high intensity, which is due to additional loss of C2H2O2 from the sugar moiety:
| Gem + e− (≈0; 0.6 eV) → C3H4F2O−(m/z 94) + C + C2H2O2 |
C3H4F2O− is formed within a broad signal peaking close to 0 eV with a clear shoulder at 0.6 eV indicating that the signal consists of contributions from different closely spaced resonances. Alternatively, the fragment ion at m/z 94 can also be assigned to the sum formula C4H3N2O− that can be formed by cleavage of the N-glycosidic bond and excision of the NH2 group and with the excess electron residing on the cytosine subunit. A similar fragment was previously observed from isolated cytosine, but at higher energies (5.2 eV and 7.3 eV).31
At m/z 115 another fragment ion is formed from all three investigated molecules that can be ascribed to N-glycosidic bond cleavage, formation of F2/H2 from the sugar ring and charge retention on the remaining sugar moiety. The corresponding ion yield curves are displayed in Fig. 4b, and all three curves show a narrow peak around zero eV and an additional signal from 0.5–1.5 eV (peaking at 1 eV from dCyt, at 1.1 eV from dFCyt and at 0.9 eV from Gem). The same fragment anion was observed in electron attachment to 2′-deoxyribose with similar resonant features close to 0 eV and 1.5 eV.25 This confirms that this fragmentation reaction is confined to the sugar moiety. It is important to emphasize that in this case also the 1 eV resonance is attributed to electron attachment to the sugar moiety (thus corresponding to a σ* resonance). Also this fragment anion is observed with higher intensity from Gem.
 | ||
| Fig. 4 (a) Reaction scheme showing the N-glycosidic bond cleavage and formation of neutral H2 and F2, respectively, from the deoxyribose (dR) ring. The charge remains on the sugar ring and thus the anion has the same m/z for all compounds investigated here. (b) Corresponding ion yield curves for m/z 115. | ||
In all three molecules a series of small fragment anions (CN−, OH−, O−/NH2− and H−) is formed from different resonances at a variety of energies. While the first two fragment anions from all investigated molecules are generated in the low energy domain, more specifically at around 2 eV (CN−) and 0.3 eV (OH−), the H− anion is generated in the high energy range between 4.5 and 8 eV (not shown). In Fig. 5 the ion yield curves of the fragment anion at m/z 16 is displayed, which can be attributed to the isobaric anions O−/NH2−. They arise from a series of higher-lying core-excited resonances. These TNIs are formed when the incoming electron induces electronic transition and is concomitantly trapped in the field of electronically excited molecule. The first resonance between 6 and 8 eV matches the energy of the first resonance of O− reported from 2′-deoxyribose.25 On the other hand, the second resonance visible between 8 and 11 eV reflects the energy of resonances observed in both components namely 2′-deoxyribose and cytosine.25,29 Thus it is very likely that both the O− and the NH2− anion contribute to the intensity of the second resonance and arise from both sub-units.
 | ||
| Fig. 5 Ion yield curves of O−/NH2− formed from dCyt, dFCyt and Gem. The signal between 6 and 8 eV is due to electron attachment to the sugar moiety and the signal between 8 and 11 eV is due to electron attachment to both sugar and cytosine moiety. | ||
Conclusions
In summary, we have investigated how fluorination of the nucleoside dCyt modifies the formation of TNIs and their subsequent decay by dissociation. In particular we compared fluorination at the cytosine base with fluorination at the 2′-deoxyribose unit, the latter molecule being an established radiosensitizer in tumor radiation therapy (where it is referred to as gemcitabine). In general we can conclude that fluorination results in a selective increase of the fragmentation rates which is ascribed to an enhancement of the corresponding electron attachment cross section. The modification of the DEA cross sections is selective in a sense that the resonances associated with the fluorinated site appear stronger without modifying the resonance energies. Specifically, for the formation of the dehydrogenated nucleobase anion we determined an enhancement factor of 2.8 upon fluorination of the sugar, whereas the enhancement factor is 5.5 when the nucleobase is fluorinated. In the case of gemcitabine additional fragmentation pathways are opened that are not accessible in 2′-deoxycytidine or 2′-deoxy-5-fluorocytidine. This may additionally contribute to the efficiency of gemcitabine as a radiosensitizer. Furthermore, the present results demonstrate that certain reactions can be controlled and enhanced by fluorination of specific molecular subunits. The present study thus represents a starting point for a molecular and physico-chemical understanding of radiosensitization by therapeutic agents, thereby enabling a rational design of next-generation radiosensitizers. For instance, a fluorination of both the sugar and the nucleobase moiety in a nucleoside will lead to more efficient decomposition by secondary low-energy electrons. However, further studies have to be performed to study the effect of incorporation of such radiosensitizers into DNA oligonucleotides on the DNA strand break yields.Acknowledgements
This work was supported by the Polish Ministry of Science and Higher Education. I.B. and A.K. acknowledge support for a visit to Siedlce, Poland, from the European Union via the COST Action MP1002 (Nano-scale insights in ion beam cancer therapy, Nano-IBCT). I.B. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG), support through a Marie Curie FP7 Integration Grant within the 7th European Union Framework Programme and from the University of Potsdam. A.K. acknowledges financial support from the Alexander von Humboldt foundation.Notes and references
- T. Y. Seiwert, J. K. Salama and E. E. Vokes, Nat. Clin. Pract. Oncol., 2007, 4, 86–100 CrossRef CAS PubMed.
- G. D. Wilson, S. M. Bentzen and P. M. Harari, Semin. Radiat. Oncol., 2006, 16, 2–9 CrossRef PubMed.
- D. S. Shewach and T. S. Lawrence, J. Clin. Oncol., 2007, 25, 4043–4050 CrossRef CAS PubMed.
- Y. Zheng, D. J. Hunting, P. Ayotte and L. Sanche, Phys. Rev. Lett., 2008, 100, 4 Search PubMed.
- L. Sanche, Chem. Phys. Lett., 2009, 474, 1–6 CrossRef CAS PubMed.
- M. Rezaee, E. Alizadeh, D. Hunting and L. Sanche, Bioinorg. Chem. Appl., 2012, 9 Search PubMed.
- H. Abdoul-Carime, M. A. Huels, E. Illenberger and L. Sanche, J. Am. Chem. Soc., 2001, 123, 5354–5355 CrossRef CAS.
- I. Baccarelli, I. Bald, F. A. Gianturco, E. Illenberger and J. Kopyra, Phys. Rep., 2011, 508, 1–44 CrossRef CAS PubMed.
- E. Alizadeh and L. Sanche, Chem. Rev., 2012, 112, 5578–5602 CrossRef CAS PubMed.
- R. Panajotovic, F. Martin, P. Cloutier, D. Hunting and L. Sanche, Radiat. Res., 2006, 165, 452–459 CrossRef CAS PubMed.
- F. Martin, P. D. Burrow, Z. L. Cai, P. Cloutier, D. Hunting and L. Sanche, Phys. Rev. Lett., 2004, 93, 4 Search PubMed.
- B. Boudaiffa, P. Cloutier, D. Hunting, M. A. Huels and L. Sanche, Science, 2000, 287, 1658–1660 CrossRef CAS.
- M. A. Huels, B. Boudaiffa, P. Cloutier, D. Hunting and L. Sanche, J. Am. Chem. Soc., 2003, 125, 4467–4477 CrossRef CAS PubMed.
- M. Rezaee, D. J. Hunting and L. Sanche, Int. J. Radiat. Oncol., Biol., Phys., 2013, 87, 847–853 CrossRef CAS PubMed.
- J. Kopyra, C. Koenig-Lehmann, I. Bald and E. Illenberger, Angew. Chem., Int. Ed., 2009, 48, 7904–7907 CrossRef CAS PubMed.
- J. Reedijk, Eur. J. Inorg. Chem., 2009, 1303–1312 CrossRef CAS.
- Y. Park, K. Polska, J. Rak, J. R. Wagner and L. Sanche, J. Phys. Chem. B, 2012, 116, 9676–9682 CrossRef CAS PubMed.
- S. Denifl, P. Candori, S. Ptasinska, P. Limao-Vieira, V. Grill, T. D. Mark and P. Scheier, Eur. Phys. J. D, 2005, 35, 391–398 CrossRef CAS.
- H. Abdoul-Carime, M. A. Huels, E. Illenberger and L. Sanche, Int. J. Mass Spectrom., 2003, 228, 703–716 CrossRef CAS.
- S. Ptasinska, S. Denifl, S. Gohlke, P. Scheier, E. Illenberger and T. D. Mark, Angew. Chem., Int. Ed., 2006, 45, 1893–1896 CrossRef CAS PubMed.
- I. Bald, I. Dabkowska and E. Illenberger, Angew. Chem., Int. Ed., 2008, 47, 8518–8520 CrossRef CAS PubMed.
- I. Anusiewicz, J. Berdys-Kochanska and J. Simons, J. Phys. Chem. A, 2005, 109, 5801–5813 CrossRef CAS PubMed.
- J. Kopyra, Phys. Chem. Chem. Phys., 2012, 14, 8287–8289 RSC.
- I. Bald, J. Langer, P. Tegeder and O. Ingolfsson, Int. J. Mass Spectrom., 2008, 277, 4–25 CrossRef CAS PubMed.
- S. Ptasinska, S. Denifl, P. Scheier and T. D. Mark, J. Chem. Phys., 2004, 120, 8505–8511 CrossRef CAS PubMed.
- I. Bald, J. Kopyra and E. Illenberger, Angew. Chem., Int. Ed., 2006, 45, 4851–4855 CrossRef CAS PubMed.
- P. Sulzer, S. Ptasinska, F. Zappa, B. Mielewska, A. R. Milosavljevic, P. Scheier, T. D. Mark, I. Bald, S. Gohlke, M. A. Huels and E. Illenberger, J. Chem. Phys., 2006, 125, 044304 CrossRef PubMed.
- T. Sommerfeld, J. Chem. Phys., 2007, 126 Search PubMed.
- S. Denifl, S. Ptasinska, M. Probst, J. Hrusak, P. Scheier and T. D. Mark, J. Phys. Chem. A, 2004, 108, 6562–6569 CrossRef CAS.
- I. Bald, J. Kopyra, I. Dabkowska, E. Antonsson and E. Illenberger, J. Chem. Phys., 2007, 126, 074308 CrossRef PubMed.
- M. A. Huels, I. Hahndorf, E. Illenberger and L. Sanche, J. Chem. Phys., 1998, 108, 1309–1312 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |