 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomimetic 3D hydroxyapatite architectures with interconnected pores based on electrospun biaxially orientated PCL nanofibers
Zhiqiang
Su
ab,
Jingfeng
Li
ac,
Zhaofei
Ouyang
a,
Matthias M. L.
Arras
b,
Gang
Wei
*c and
Klaus D.
Jandt
*b
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China
bChair of Materials Science, Otto Schott Institute of Materials Research, Friedrich-Schiller-University Jena, D-07743 Jena, Germany. E-mail: k.jandt@uni-jena.de
cHybrid Materials Interfaces Group, Faculty of Production Engineering, University of Bremen, D-28359 Bremen, Germany. E-mail: wei@uni-bremen.de
First published on 13th March 2014
Abstract
We report here a facile strategy to fabricate three-dimensional (3D) hydroxyapatite (HA) architectures with well-defined long continuous interconnected pores by using electrospinning and biomimetic mineralization. To this end, a polymeric nanofiber (NF) scaffold with well-defined architecture was fabricated by electrospinning, and bone morphogenetic protein 2 (BMP2) was then adsorbed onto the chemically modified NFs through bio-conjugation. The 3D nanoporous HA architecture was finally fabricated by biomimetic mineralization of the NF–BMP2 hybrid in simulated body fluids and subsequent dissolution of NFs in hexafluoroisopropanol. The formation of NF–BMP2 hybrid was identified by confocal laser scanning microscopy analysis. The crystal structure of HA crystals formed on NFs was examined by X-ray diffraction. The chemical composition and interconnected porous structure of the created 3D HA architectures were measured by X-ray photoelectron spectroscopy, focused ion beam scanning electron microscopy, and transmission electron microscopy, respectively. This bottom-up strategy based on electrospinning and biomimetic mineralization opens up a new way to prepare diverse porous HA-based hybrid materials and shows great potential in drug delivery, gene transfer and tissue engineering.
1. Introduction
Hydroxyapatite (HA) is an important inorganic material in the hard tissues of bone and teeth, and it is one of the few materials that can bind to bone tissue and promote bone substitution and repair.1,2 There is increasing interest in the preparation and application of novel HA-based composite materials for bone substitutes.3–5 It is well known that natural bone is porous and mainly composed of highly aligned HA crystals that bind onto the arranged type I collagen matrix.6 To mimic the structure of natural bone, various strategies, such as polymeric sponging,7 rapid prototyping,8 phase separation,9 salt leaching,10 incorporation of porogen particles,11 freeze casting,12 and gel-casting,13 have been used to fabricate porous HA-based biomaterials.Porous HA-based materials have been used in drug delivery, dental surgery, and protein carrying.14–16 The pore size and structure of the porous HA materials are important factors for the formation of new bone substitutes.17 Until now, only few studies have reported the substitution of newly-formed bone with the implanted HA-based materials,18 and a main reason is probably that the HA material has closed structures lacking interconnected pores.19 Therefore, the fabrication of HA-based materials with highly interconnected pores, bioactivity as well as good mechanical strength is required for maximizing their functions and in vivo applications. Up to now, it still remains a challenge to prepare interconnected porous HA scaffolds with high mechanical strength because sometimes they are so brittle.
Electrospinning is a simple and highly versatile technique for the large-scale preparation of polymeric nanofibers (NFs) with uniform diameter ranging from 20 nm to hundreds μm,20,21 and it has been widely applied in the biomedical field.22–24 After the creation of polymeric NFs, the coating of HA crystals can be performed by biomineralization in simulated body fluid (SBF).25 To promote the nucleation and growth of HA, various functional groups (such as carboxyl, hydroxyl and amino groups) and biomolecules (fibrinogen, fibronectin, collagen, chitosan, gelatin, heparin, etc.) have been introduced to modify the surface of polymeric NFs. With this strategy, Liu et al. prepared HA materials with 1D hollow tubular structures.25 They first treated the electrospun poly(lactic-co-glycolic acid) (PLGA) fibers with plasma, chitosan and heparin successively, and realized a coating of HA by immersing the modified nanofibers into a modified 10-fold concentrated SBF. Further, by dissolving the PLGA NFs in the core, 1D hollow tubular HA materials were created. Recently, we have developed a method to prepare uniaxially and biaxially oriented electrospun fiber mats, and with our improved electrospun apparatus fiber mats with designed fiber assembly and controlled fiber spacing have been obtained.26,27 Based on our previous studies and inspired by Liu's report,25–27 we developed a new bottom-up strategy to prepare 3D HA architectures with well-defined long continuous interconnected pores by using electrospinning and biomimetic mineralization. Electrospinning is superior to other techniques not only in its easy functionalization of the as-spun NFs with various biomolecules, such as proteins, peptides and amino acids but also in its controllability for fabricating various patterns of NFs, such as the random, axially and biaxially aligned NFs and well-designed 3D fibrous scaffolds.26–29
In this work, electrospinning was utilized to create 3D biaxially orientated polymer NFs and bone morphogenetic protein 2 (BMP2) was adsorbed onto the polymer NFs to mediate the biomineralization. HA was formed by immersing the NF–BMP2 architectures in SBF for at least 7 days, and the interconnected pores in HA architectures were obtained by dissolving the NF templates. The obtained 3D HA architectures were characterized and a possible formation mechanism of HA crystals on NFs was proposed. The interconnected porous structure of the created 3D HA material was characterized by microscopy analysis. To the best of our knowledge, it is the first report to create interconnected porous 3D HA architectures with the electrospun biaxially oriented polymer NF scaffolds. In addition, we utilized a biocompatible bone protein (BMP2) to improve the efficiency of biomimetic mineralization, and HA crystals were formed on NF–BMP2 architectures in normal SBF solution (instead of 10-fold-concentrated SBF in ref. 25) quickly.
2. Experimental section
2.1. Materials
Poly (ε-caprolactone) (PCL, 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), hexafluoroisopropanol (HFIP), 1,6-hexanediamine, glutaraldehyde, BMP2, fluorescein isothiocyanate (FITC), acetic acid, and all the chemicals for the preparation of SBF were obtained from Sigma-Aldrich. All of them were used as received without any purification.
000 g mol−1), hexafluoroisopropanol (HFIP), 1,6-hexanediamine, glutaraldehyde, BMP2, fluorescein isothiocyanate (FITC), acetic acid, and all the chemicals for the preparation of SBF were obtained from Sigma-Aldrich. All of them were used as received without any purification.
2.2. Fabrication of PCL NF scaffold
The solution for electrospinning was prepared by dissolving PCL pellets in HFIP at a concentration of 8 wt%. The solution was loaded into a 10 mL plastic syringe with a 20 gauge blunt tip needle and was dispensed at a rate of 0.8–1.0 mL h−1 during electrospinning. The working distance between the tip of the needle and the collector was ∼15 cm and a voltage of 15–20 kV was applied. Alignment of the PCL NF was achieved by electrospinning with the aid of parallel placed auxiliary electrodes and a high speed rotating mandrel collector. The two auxiliary electrodes were equally charged at a voltage of 12–15 kV. The linear velocity of the mandrel collector was fixed at ∼10 m s−1. The biaxially oriented NF mesh was prepared by electrospinning multiple layers in a 0°/90° lay-up.2.3. Adsorption of BMP2 onto PCL NFs
BMP2 was immobilized onto the PCL NF surface via an aminolysis method according to a previous report.30 Firstly, the PCL NF meshes were immersed in 1,6-hexanediamine solution with suitable concentrations for 10 h at 37 °C, rinsed with deionized water for 24 h at room temperature to remove free 1,6-hexanediamine, and dried in a vacuum at 30 °C for 24 h to constant weight. Then, the aminolyzed PCL NF meshes were immersed in 1 wt% glutaraldehyde solution for 3 h at room temperature, followed by rinsing with a large amount of deionized water for another 24 h to remove free glutaraldehyde. Afterwards, the NF meshes were incubated in 0.01 mg mL−1 BMP2 solution (pH = 7) for 24 h at 2–4 °C. Finally the BMP2-immobilized NF meshes were rinsed with 1.0% acetic acid solution and then rinsed with deionized water for 24 h to remove free BMP2.2.4. Fabrication of 3D HA architecture
Biomimetic mineralization of BMP2-coated NF scaffolds was performed according to our previous reports.31,32 In brief, the BMP2-coated PCL NF meshes were incubated in 25 mL of a SBF solution (142 mM Na+, 5.0 mM K+, 1.5 mM Mg2+, 2.5 mM Ca2+, 152.0 mM Cl−, 4.2 mM HCO3−, 1.0 mM HPO42−, and 0.5 mM SO42−, buffered at pH 7.4 with Tris–HCl) at 37 °C for bone-like mineral growth. All the fluids were renewed every day. The samples were taken out at different time intervals and rinsed by large amounts of water to remove the excessively adsorbed salts, and then dried in air for characterization. The mineralized NF–HA hybrids were immersed in HFIP for 48 h to remove the PCL component, then rinsed with deionized water and dried in air at room temperature.2.5. Characterization techniques
The morphologies of the PCL and HA samples were examined by focused ion beam scanning electron microscopy (FIB-SEM, AURIGA 60, Carl-Zeiss Company, Germany). To visualize the internal pore structure of the final 3D nanoporous HA architecture, FIB was utilized to cross-section the sample. Before the sectioning process, a 500 nm thick Pt layer was deposited on the sample to protect the surface layers from ion beam damage. A large trench was milled at 30 kV and an ion beam current of 2 nA for 60 s. The fluorescence images of PCL NF meshes coated with FITC labeled BMP2 were taken using a LSM Meta 510 confocal laser scanning microscopy (CLSM, Carl-Zeiss, Germany). Optical excitation was carried out with a 488 nm argon-ion laser beam and fluorescence emission was detected at 520 nm. X-ray diffraction (XRD) was carried out using a Rigaku D/max-2500 X-ray Diffractometer with CuK radiation (k = 0.15406 nm) operating at 40 kV and 200 mA. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a PHI Quantum 2000 spectrometer (PHI Co., Chanhassen, MN, USA) to analyse the HA crystals formed on the NF–BMP2 architectures, in order to determine the probable formation time of HA. A monochromatic Al Kα radiation as the X-ray source (hν = 1486.6 eV) was employed and a 100 μm diameter beam was used. The electron take-off angle was 45° and the analyzer was operated in the constant energy mode for all measurements. Transmission electron microscope (TEM) experiments were performed on a JEOL 3010 electron microscope (Tokyo, Japan) with an accelerating voltage of 300 kV. The mineralized HA architecture was cut to small pieces and dispersed with ultrapure water, and then dropped onto a Cu grid for TEM characterization.3. Results and discussion
Fig. 1 schematically shows the main steps for preparing 3D porous HA architectures by using electrospinning and biomimetic mineralization. Firstly, a PCL NF scaffold with well-defined architecture is fabricated through electrospinning, and then the as-spun PCL NFs are chemically modified and coated with BMP2 via bio-conjugation (Step 1). Secondly, the BMP2-coated PCL NFs are mineralized in SBF to form HA crystals along the NFs through the mediation of BMP2 proteins (Step 2). Finally, PCL NFs are removed by immersing the mineralized materials in HFIP (Step 3). In this way, 3D porous HA architectures with interconnected pores are created. | ||
| Fig. 1 Schematic model for fabricating 3D porous HA architecture. | ||
3.1. Electrospinning NF architectures for BMP2 adsorption
We achieved well oriented PCL NF architectures by using our home-made electrospinning set-up which incorporates a pair of parallel auxiliary electrodes, a high speed rotating mandrel collector and a mobile platform (Fig. 2a). Detailed description can be found in our previous studies.27 The parallel positioned auxiliary electrodes can symmetrically reduce the bending instability of the polymer jet, so that aligned and straight NFs are deposited on a high speed rotating mandrel collector. The mobile platform that allows the spinneret to move left and right at an adjustable rate controls the placement of electrospun NFs. For different applications, the NF meshes can be prepared with adjustable size and uniform woven structure (Fig. 2b). | ||
| Fig. 2 (a) Photograph of our home-made electrospinning apparatus and (b) optical image of electrospun PCL NF mesh with a macroscopic size. (c and d) FIB-SEM images of electrospun NFs with uniaxial (c) and biaxial (d) orientations. | ||
With our improved home-made electrospinning apparatus, polymer NFs with different orientations (uniaxially and biaxially) and various layers can be prepared easily. Fig. 2c and d show the FIB-SEM images of uniaxially and biaxially aligned PCL NF meshes, respectively. The created PCL NFs are uniform and the average diameter is about 100–200 nm.
In our previous work, we found that some proteins, such as fibrinogen and fibronectin, can promote the nucleation and growth of HA nanocrystals and microflakes.31,32 The carboxylic groups on the protein surface may provide enough binding sites for Ca2+ and hence the protein templates can serve as scaffolds for biomimetic mineralization.33,34 BMP2 is an osseoinductive protein that induces new host bone formation by fluiding the modulation and promoting differentiation of mesenchymal cells into bone forming cells.35–38 Due to its activity in bone repair and formation, BMP2 was utilized to bind onto the as-spun PCL NFs for biomineralization in this work.
CLSM was used to confirm the immobilization of BMP2 onto PCL NFs. Pure PCL NF architectures were examined as control (Fig. 3a and b), and there is very weak fluorescence for both uniaxially and biaxially aligned NFs. However, after conjugation with FITC-labelled BMP2, it can be found that the fluorescence of PCL NFs increased greatly (Fig. 3c and d), which indicates the successful coating of BMP2 onto the surface of PCL NFs.
 | ||
| Fig. 3 CLSM images of uniaxially and biaxially aligned NF architectures before (a and b) and after (c and d) coating with BMP2. | ||
3.2. Growth of HA on NF–BMP2 architectures
After the modification of NFs with BMP2 protein, the created NF–BMP2 architectures were incubated in SBF to form HA crystals. SEM was utilized to observe the change of morphologies of apatite on NF–BMP2 architectures with the increase of the incubation period, and the corresponding images are shown in Fig. 4. The minerals started to grow along the NFs after 1 day incubation, and globular apatite nanoparticles were formed along the NFs (Fig. 4a). After 3 days incubation, the globular nanoparticles grew on one hand in the direction perpendicular to the NFs and evolved into needlelike minerals, and on the other hand, they also spread along the NFs and gradually connected with each other and finally formed tabular microcrystals (Fig. 4b). While after 7 days' incubation the minerals have almost spread all over the fiber mesh, leaving the fibres vaguer (Fig. 4c). With the incubation period increasing to 14 days, it was observed from the SEM images that the surfaces of NF architectures were fully covered with minerals (Fig. 4d). | ||
| Fig. 4 FIB-SEM characterization of apatite crystals formed on BMP2-modified biaxially aligned NF architectures in SBF with different incubation periods: (a) 1, (b) 3, (c) 7, and (d) 14 days. | ||
To confirm the minerals formed around the NF surfaces are apatite, XRD characterization was conducted. Fig. 5 shows the XRD patterns of the NF architectures after soaking in SBF for different periods of time. The XRD pattern of NF architecture after soaking for 1 day shows a broad peak between 30° and 35°, with a maximum at about 32°, corresponding to the apatite (002) diffraction peak.25,39,40 A slight sharpening of the broad peak is observed in the XRD pattern of the NF architecture after soaking for 3 days. Further sharpening of the peak is observed in the XRD patterns of the architecture soaked for longer periods (7 and 14 days). The (211) diffraction peak becomes obvious after 3 days soaking and became very sharp in the XRD patterns of scaffolds soaked for 7 and 14 days. Other diffraction apatite peaks (102, 222, 213, and 004) also become more evident after the architecture is soaked for longer periods (7 and 14 days). Therefore, it can be concluded that the biomimetic formation of HA is successful and HA crystals were finally formed after at least 7 days incubation in SBF.
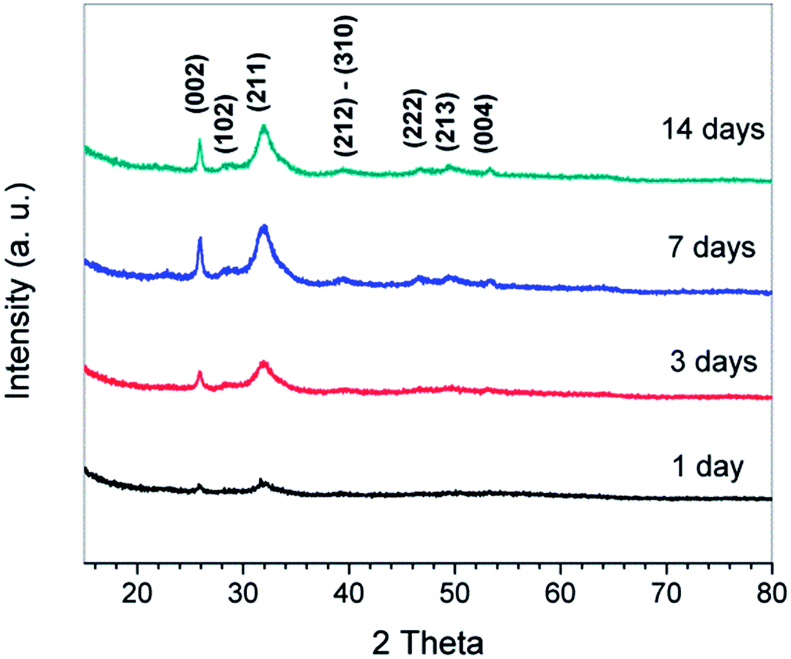 | ||
| Fig. 5 XRD patterns of the fiber scaffold after soaking in SBF for different periods of time. | ||
To determine the atomic ratio of Ca/P and further ascertain the formation of apatite, the fiber architectures incubated in SBF for different periods of time were further measured by XPS. In a typical XPS spectrum, the peaks of Ca 2p (347.5 eV), P 2s (191.3 eV) and P 2p (132.8 eV) can be clearly seen (Fig. 6), which is in agreement with the previously reported data.31 It should be noted that the elements measured by XPS are located on the surface of apatite crystals since the detection depth of X-ray source of the XPS instrument is about 6–9 nm. The Ca/P ratio increased with the on-going of biomineralization process (inset of Fig. 6), and this phenomenon has been documented in earlier reports.31,41,42 The Ca/P ratio of the initial minerals formed at the beginning of mineralization is about 1.33, which indicates that a Ca-poor amorphous calcium phosphate (ACP) was formed on the NF–BMP2 hybrids.39 With the increasing of incubation period, Ca/P ratios of 1.47, 1.58 and 1.66 are found after 3, 7 and 14 days of biomimetic mineralization, respectively (see the inset), suggesting the gradual recrystallization of ACP into apatites, which is in good agreement with the XRD result. The Ca/P ratio of the mineralized NF–BMP2 hybrids at 14 days was slightly lower than that of stoichiometric HA (1.67) due to the incorporation of Na, Mg and carbonate. This indicates that the apatite produced on NF–BMP2 hybrids in SBF is a biological HA, which has been typically observed in those minerals created by biomimetic mineralization.41,43
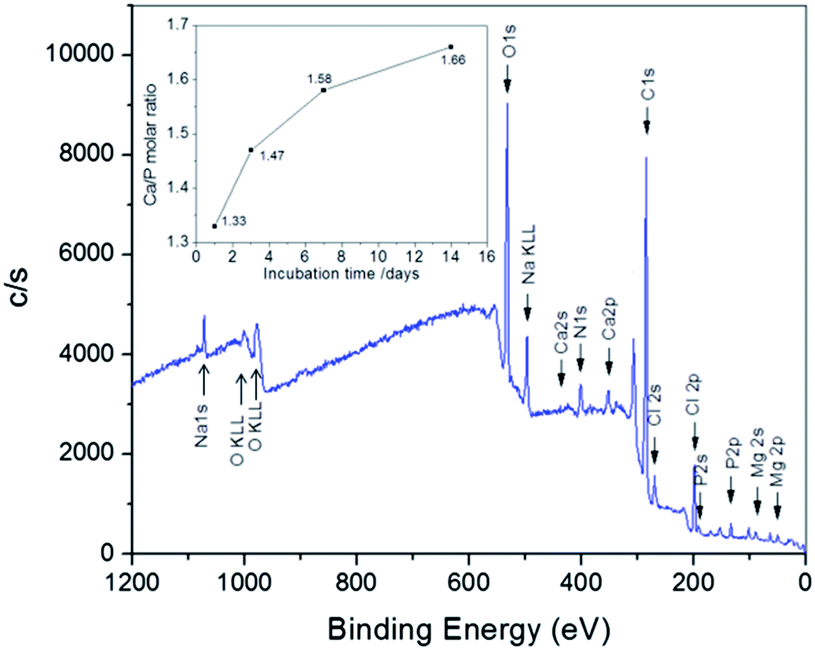 | ||
| Fig. 6 Typical XPS spectrum of apatite minerals formed in SBF after 7 days. The inset gives the atomic Ca/P ratio of obtained apatite architectures with different mineralization periods. | ||
Based on these XPS data, we suggest that BMP2 can promote the nucleation and growth of HA crystals and 3D HA architectures can be created by incubating BMP2-modified NF meshes in SBF for about 7 days. In a previous study, Li et al. reported the creation of silk–BMP2 scaffolds by electrospinning and further investigated their application in bone tissue engineering.37 They found that the scaffolds with the co-processed BMP2 supported higher calcium deposition and enhanced transcript levels of bone-specific markers than the control scaffolds without BMP2 and, therefore, could be used as potential candidate for bone tissue engineering.
3.3. Possible nucleation mechanism
To understand the mineralization mechanism mediated by BMP2, control experiments were performed by investigating the biomineralization of BMP2-modified uniaxially aligned PCL NFs in SBF. A previous mineralization experiment on biaxial PCL nanofibers (Fig. 4) indicates that the formation of tubular HA microcrystals is related to three factors, i.e. the grids of nanofiber mesh, the proteins, and the axis of nanofibers, therefore we utilized the uniaxial electrospun nanofiber mesh for the biomineralization of the HA architecture. It can be found from the SEM images that the precipitates were also formed firstly along the NFs and then grew perpendicularly to the NFs as well as laterally along the NFs to connect with each other to form tabular HA microcrystals, and finally covered the NFs, as shown in Fig. 7. The mineralization process is similar to that on the biaxially aligned PCL NFs (Fig. 4). Therefore, it can be concluded that the tubular HA microcrystals are not related to the electrospun nanofiber structures but to the BMP2 protein and the perpendicular axis of nanofibers. | ||
| Fig. 7 FIB-SEM images of apatite crystals on BMP2-modified uniaxially aligned PCL NFs with different incubation periods: (a) 1, (b) 3, (c) 7, and (d) 14 days. | ||
Based on the obtained SEM (Fig. 4 and 7), XRD (Fig. 5) and XPS (Fig. 6) results, we propose a possible mechanism for the nucleation and growth of HA on NF–BMP2 architectures, as shown in Fig. 8. After exposing the NF–BMP2 to SBF, globular ACP were formed along the NFs within 1 day (Step 1). It is clear that the BMP2 molecules provide binding sites for Ca2+ and can promote the nucleation and growth of apatite nanocrystals, which agrees well with our previous studies on biomineralization of protein NFs.31,32 With the increase of the exposing period to 3 days, more Ca2+ ions were adsorbed onto the surface of the globular ACP precursors and the precipitates grew vertically to form needlelike crystals as well as laterally to connect with each other and finally evolved into tabular minerals (Step 2). In this process, the NF templates control the growth of minerals in the direction parallel to the NFs, while BMP2 controls the growth of created ACP in the direction perpendicular to the NFs, which is similar to the formation of HA on carbon nanotube.43 After 7 and 14 days incubation in SBF, the formed tabular ACP recrystallized and grew into a larger piece HA and finally covered the NFs to form 3D architectures (Step 3).
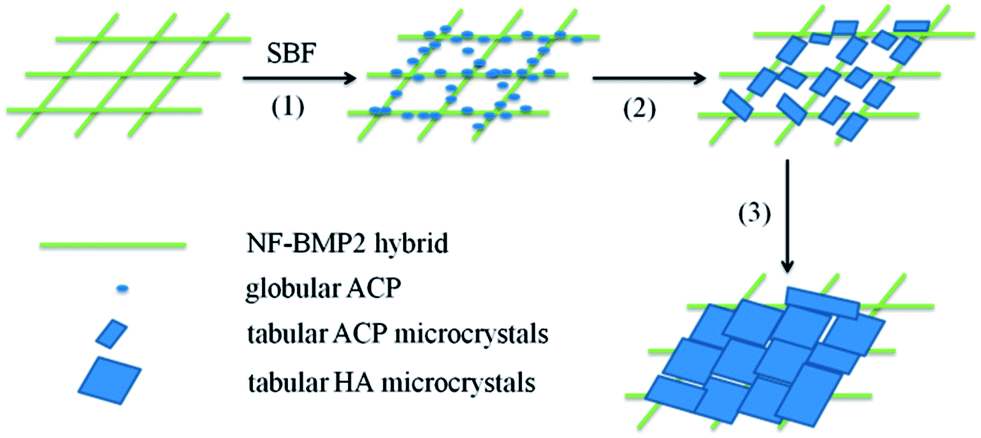 | ||
| Fig. 8 Proposed mineralization mechanism of HA on NF–BMP2 hybrids. | ||
3.4. Fabrication of 3D HA architectures with interconnected pores
Although HA-based porous materials have been widely used in bone tissue engineering, previous biological studies and clinical practices have identified that 3D interconnected porous structures are necessary to allow cell attachment, proliferation, and differentiation, and to provide pathways for biofluids and blood vessels.7,17–19 Here we achieved the creation of 3D HA architectures with interconnected pores by removing the PCL substrate from the NF–BMP2–HA bulk materials with HFIP solution.In order to create a 3D HA-based porous architecture, a woven biaxially aligned PCL NF architecture with 8 layers (Fig. 9a) was used as a template to bind with BMP2 and the obtained hybrid material was mineralized in SBF for 14 days, and the final 3D NF–BMP2–HA bulk materials were prepared (Fig. 9b). A 3D porous HA architecture was obtained by removing the PCL substrate with HFIP solution. To verify its internal interconnected porous structure, the porous HA material was cross-sectioned by focused ion beam (FIB) of the FIB-SEM. The FIB was used as a nano-scalpel to perform serial sectioning on the sample and create a 3D image of the internal structure of the sample. Fig. 9c and d show the 3D SEM images of nanoporous HA architecture (height > 2 μm) after cross-section milling with different sample rotations of 54° and 0°, respectively. It can be clearly seen that the precursor PCL NFs were successfully removed, leaving criss-crossing grooves and pores (indicated as arrows) embedded in the as-milled surface, which confirms the long continuous interconnected pore structure of the created 3D HA architecture. In a control experiment, the uniaxial PCL NF mesh was utilized for the mineralization of HA architecture. After incubation in SBF solution for 14 days, the bulk 3D HA architecture was created on the uniaxial NF mesh (Fig. 9e). After milling with FIB, the created HA architecture shows some uniaxial grooves and no obvious interconnected pores are found in this architecture (Fig. 9f). Therefore, the advantage of our biaxial PCL NF mesh to the uniaxial NF mesh for the fabrication of interconnected porous HA architecture is proved.
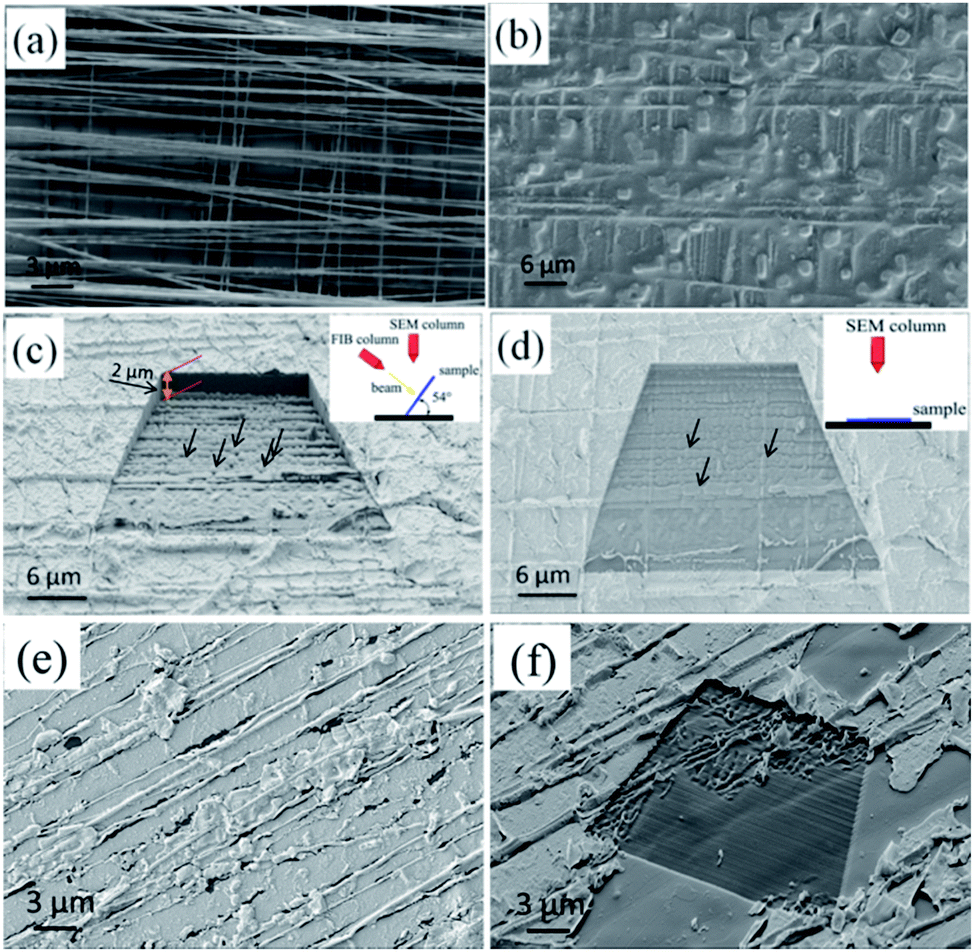 | ||
| Fig. 9 FIB-SEM images of woven biaxial PCL NF mesh with 8 layers (a) before and (b) after mineralization in SBF. (c and d) 3D SEM images of porous HA architecture after milling with FIB under a sample rotation of (c) 54° and (d) 0°, respectively. (e and f) FIB-SEM and corresponding milling 3D SEM images of HA architecture with uniaxial PCL NF mesh. The apparent grooves reveal the successful removal of PCL NFs in the bulk. The black arrows indicate the pores caused by NF removing. | ||
To further prove the interconnected porous structure of 3D HA architecture formed on biaxial PCL NF mesh, TEM was carried out. Fig. 10a shows the typical TEM image of 3D porous HA architecture deposited on a Cu grid. It is obvious that our HA material has an interconnected porous structure. A statistical analysis of the pore size indicates that the size of the pores is about 400–900 nm (Fig. 10b). This pore size is much larger than that of the diameter of NFs (100–200 nm). We suggest that maybe some HA particles were removed during the ultrasonic treatment process for the TEM sample preparation, or some of the electrospun NFs were aggregated, causing the increase of the pore size.
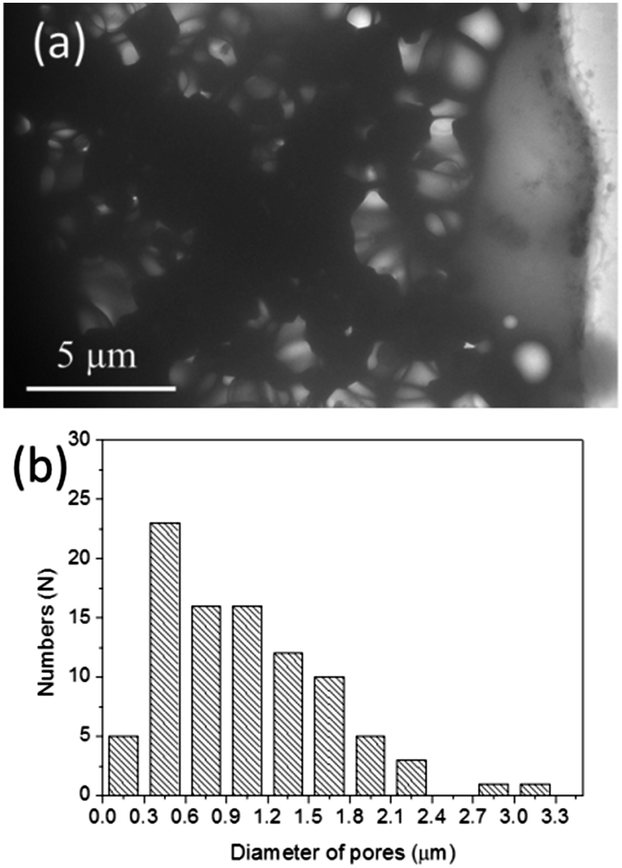 | ||
| Fig. 10 (a) Typical TEM image of the fabricated interconnected porous HA 3D architecture, and (b) corresponding size distribution of pores. | ||
Basically, this porous structure provides pathways for biofluids and it is essential for nutrient transport, protein and gene transfer. Furthermore, the bioactive BMP2 layer persisting on the internal surface of these pores can further endow the porous apatites more functions such as promoting the cell adhesion, proliferation, and differentiation. Admittedly as a bone repair material, larger pore size is preferable. In next step, we plan to create 3D interconnected porous HA-based materials with hierarchical pore size of macro- to nanoscale by adjusting the electrospinning parameters and combining it with the melt electrospinning method, which is capable of producing thicker fibers.44 And in the follow-up research we also plan to further evaluate the mechanical strength and bioactivity of this 3D HA porous material and examine whether the BMP2 is still left on the surface of the internal pores and its effect on the cell adsorption and growth.
4. Conclusions
In summary, we present a new bottom-up strategy for the preparation of 3D HA architectures with well-defined long periodic interconnected pores by using electrospinning and biomimetic mineralization. We found that PCL NFs and BMP2 had strong effects on the nucleation and growth of HA crystals, and both of them are necessary for the creation of 3D porous HA materials. The adsorbed protein molecules promote the binding of Ca2+ onto the NFs and template the vertical growth of HA minerals and the electrospun NFs guide the lateral formation of HA crystals. Based on the obtained results, we propose a possible mechanism for the formation of apatite crystals mediated by BMP2. The bottom-up strategy based on electrospinning and biomineralization shown in this work opens up a new way to prepare diverse porous HA-based hybrid materials, since NF meshes with various outlines and fiber assemblies can be achieved by electrospinning and various bioactive molecules can be bound onto the NF meshes through surface modification to generate more biofunctions. The fabricated 3D porous HA architectures will have potential applications in drug delivery, protein and gene transfer.Acknowledgements
Z. S. and K. D. J. gratefully acknowledge the partial financial support of the Alexander von Humboldt Foundation, Bonn, Germany in the form of a Humboldt Research Fellowship for Experienced Researchers, the Fundamental Research Funds for the Central Universities (Project no. ZZ1307), the Beijing New-Star Program of Science and Technology (2009B10), the Deutsche Forschungsgemeinschaft (DFG, grant no. INST 275/241-1 FUGG) and the Thüringer Ministerium für Bildung, Wissenschaft und Kultur (TMBWK, grant no. 62-4264 925/1/10/1/01). G. Wei and J. Li would like to thank the financial support of the China Scholarship Council (CSC) PhD scholarship.Notes and references
- A. Balamurugan, A. H. S. Rebelo, A. F. Lemos, J. H. G. Rocha, J. M. G. Ventura and J. M. F. Ferreira, Dent. Mater., 2008, 24, 1374 CrossRef CAS PubMed.
- S. Weiner and H. D. Wagner, Annu. Rev. Mater. Sci., 1998, 28, 271 CrossRef CAS.
- H. Yoshikawa, N. Tamai, T. Murase and A. Myoui, J. R. Soc. Interface, 2009, 6, S341 CrossRef CAS PubMed.
- B. Palazzo, M. Iafisco, M. Laforgia, N. Margiotta, G. Natile, C. L. Bianchi, D. Walsh, S. Mann and N. Roveri, Adv. Funct. Mater., 2007, 17, 2180 CrossRef CAS.
- Z. Ge, S. Baguenard, L. Y. Lim, A. Wee and E. Khor, Biomaterials, 2004, 25, 1049 CrossRef CAS.
- P. Fratzl, H. S. Gupta and E. P. Paschalis, J. Mater. Chem., 2004, 14, 2115 RSC.
- H. R. Ramay and M. Zhang, Biomaterials, 2003, 24, 3293 CrossRef CAS.
- J. M. Williams, A. Adewunmi, R. M. Schek, C. L. Flanagan, P. H. Krebsbach, S. E. Feinberg, S. J. Hollister and S. Das, Biomaterials, 2005, 26, 4817 CrossRef CAS PubMed.
- A. R. Boccaccini and V. Maquet, Compos. Sci. Technol., 2003, 63, 2417 CrossRef CAS.
- D. Tadic, F. Beckmann, K. Schwarz and M. Epple, Biomaterials, 2004, 25, 3335 CrossRef CAS PubMed.
- D. M. Liu, J. Mater. Sci.: Mater. Med., 1997, 8, 227 CrossRef CAS.
- S. Deville, E. Saiz and A. P. Tomsia, Biomaterials, 2006, 27, 5480 CrossRef CAS PubMed.
- T. M. G. Chu, J. W. Halloran, S. J. Hollister and S. E. Feinberg, J. Mater. Sci.: Mater. Med., 2001, 12, 471 CrossRef CAS.
- W. H. Kim, J. C. Knowles and H. E. Kim, Biomaterials, 2004, 25, 1279 CrossRef PubMed.
- Y. Mizushima, T. Ikoma, J. Tanaka, K. Hoshi, T. Ishihara, Y. Ogawa and A. Ueno, J. Controlled Release, 2006, 110, 260 CrossRef CAS PubMed.
- G. Carotenuto, G. Spagnuolo, L. Ambrosio and L. Nicolais, J. Mater. Sci.: Mater. Med., 1999, 10, 671 CrossRef CAS.
- L. A. Cyster, D. M. Grant, S. M. Howdle, F. R. A. J. Rose, D. J. Irvine, D. Freeman, C. A. Scotchford and K. M. Shakesheff, Biomaterials, 2005, 26, 697 CrossRef CAS PubMed.
- T. Nakasa, O. Ishida, T. Sunagawa, A. Nkamae, Y. Yasunaga, M. Agung and M. Ochi, J. Biomed. Mater. Res., Part A, 2005, 75, 350 CrossRef PubMed.
- R. A. Ayers, S. J. Simske, C. R. Nunes and L. M. Wolford, J. Oral Maxillofac. Surg., 1998, 56, 1297 CrossRef CAS.
- L. Larrondo and S. J. Manley, J. Polym. Sci., Polym. Phys. Ed., 1981, 19, 909 CrossRef CAS.
- B. Sundaray, V. Subramanian, T. S. Natarajan, R.-Z. Xiang, C.-C. Chang and W.-S. Fann, Appl. Phys. Lett., 2004, 84, 1222 CrossRef CAS PubMed.
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151 CrossRef CAS.
- J. T. McCann, D. Li and Y. Xia, J. Mater. Chem., 2005, 15, 735 RSC.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670 CrossRef CAS PubMed.
- W. Liu, Y. C. Yeh, Y. Lipner, J. Xie, H. W. Sung, S. Thomopoulos and Y. Xia, Langmuir, 2011, 27, 9088 CrossRef CAS PubMed.
- M. M. L. Arras, C. Grasl, H. Bergmeister and H. Schima, Sci. Technol. Adv. Mater., 2012, 13, 035008 CrossRef.
- (a) Z. Su, J. Li, Q. Li, T. Ni and G. Wei, Carbon, 2012, 50, 5605 CrossRef CAS PubMed; (b) Z. Ouyang, J. Li, J. Wang, Q. Li, T. Ni, X. Zhang, H. Wang, Q. Li, Z. Su and G. Wei, J. Mater. Chem. B, 2013, 1, 2415 RSC.
- S. J. Cho, B. Kim, T. An and G. Lim, Langmuir, 2010, 26, 14395 CrossRef CAS PubMed.
- C. Grasl, M. M. L. Arras, M. Stoiber, H. Bergmeister and H. Schima, Appl. Phys. Lett., 2013, 102, 053111 CrossRef PubMed.
- Y. Zhu, C. Gao, X. Liu and J. Shen, Biomacromolecules, 2002, 3, 1312 CrossRef CAS PubMed.
- G. Wei, J. Reichert, J. Bossert and K. D. Jandt, Biomacromolecules, 2008, 9, 3258 CrossRef CAS PubMed.
- G. Wei, J. T. Zhang, L. Xie and K. D. Jandt, Carbon, 2011, 49, 2216 CrossRef CAS PubMed.
- G. K. Toworfe, R. J. Composto, I. M. Shapiro and P. Ducheyne, Biomaterials, 2006, 27, 631 CrossRef CAS PubMed.
- G. He, A. Ramachandran, T. Dahl, S. George, D. Schultz, D. Cookson, A. Veis and A. George, J. Biol. Chem., 2005, 280, 33109 CrossRef CAS PubMed.
- J. A. Koempel, B. S. Patt, K. O'Grady, J. Wozney and D. M. Toriumi, J. Biomed. Mater. Res., 1998, 41, 359 CrossRef CAS.
- E. A. Wang, V. Rosen, J. S. D'Alessandro, M. Bauduy, P. Cordes, T. Harada, D. I. Israel, R. M. Hewick, K. M. Kerns and P. LaPan, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2220 CrossRef CAS.
- C. Li, C. Vepari, H. J. Jin, H. J. Kim and D. L. Kaplan, Biomaterials, 2006, 27, 3115 CrossRef CAS PubMed.
- T. Nakasa and H. Yoshikawa, J. Bone Miner. Metab., 2006, 24, 425 CrossRef PubMed.
- M. Vallet-Regí, A. M. Romero, C. V. Ragel and R. Z. LeGeros, J. Biomed. Mater. Res., 1999, 44, 416 CrossRef.
- T. Furuzono, T. Taguchi, A. Kishida, M. Akashi and Y. Tamada, J. Biomed. Mater. Res., 2000, 50, 344 CrossRef CAS.
- H. M. Kim, T. Himeno, M. Kawashita, T. Kokubo and T. Nakamura, J. R. Soc. Interface, 2004, 1, 17 CrossRef CAS PubMed.
- J. Huang, C. Wong, A. George and D. L. Kaplan, Biomaterials, 2007, 28, 2358 CrossRef CAS PubMed.
- M. Wu, Q. Wang, X. Liu and H. Liu, Carbon, 2013, 51, 335 CrossRef CAS PubMed.
- D. W. Hutmacher and P. D. Dalton, Chem.–Asian J., 2011, 6, 44 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |