
A gel-state 2D-NMR method for plant cell wall profiling and analysis: a model study with the amorphous cellulose and xylan from ball-milled cotton linters†
Abstract
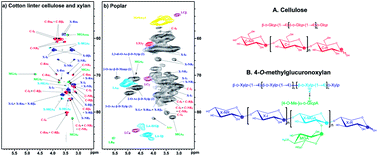
A recently developed “gel-state NMR method” that simply swells plant cell walls in a DMSO-d6/pyridine-d5 (4 : 1) solvent system and uses a high-resolution solution state 2D-NMR (HSQC) technique has been successfully applied to whole plant cell wall 2D-NMR profiling studies. However, there was limited information to assign many polysaccharide peaks unlike lignin structures. Here we collected NMR data from various cellulose and xylan models using the same solvent system to assign the unknown peaks. Furthermore, DMSO-soluble cellulose and xylan fractions were prepared from ball-milled cotton linter cellulose, and the detailed chemical structures were analyzed. The major component of cotton is cellulose (95–99%), but it typically contains ∼2% hemicelluloses. Xylan in particular was isolated and identified along with the amorphous cellulose in this study. The fully assigned spectra of cellulose and xylan provided invaluable database information for peak assignment and authentication that will be directly used to screen and identify the two main polysaccharide components from various whole cell wall NMR spectra in the same solvent system.
 Please wait while we load your content...
Please wait while we load your content...

