Dual-stimuli reduction and acidic pH-responsive bionanogels: intracellular delivery nanocarriers with enhanced release†
Yifen
Wen
and
Jung Kwon
Oh
*
Department of Chemistry and Biochemistry, Centre for NanoScience Research, Concordia University, Montreal, Quebec H4B 1R6, Canada. E-mail: john.oh@concordia.ca; Tel: +1-514-848-2424 ext. 5306
First published on 5th November 2013
Abstract
Biopolymer-based nanogels (bionanogels) are a promising platform as polymer-based drug delivery systems encapsulting hydrophilic anticancer therapeutics; however, enhanced/controlled drug release is highly desired. Herein, we report new dual stimuli-responsive bionanogels (ssBNGs) as potential intracellular delivery nanocarriers with multi-controlled and enhanced drug release. A facile aqueous crosslinking polymerization of oligo(ethylene oxide)-containing methacrylate (OEOMA) in the presence of carboxymethyl cellulose (CMC) and a disulfide-labeled dimethacrylate allows for the synthesis of ssBNGs crosslinked with disulfide linkages of POEOMA-grafted CMC. These ssBNGs exhibit dual response release of encapsulated anticancer drugs: reductive cleavage of disulfide crosslinks and acidic pH-response of carboxylic acid groups in CMC. Their applicability toward tumor-targeting drug delivery applications is demonstrated with confocal laser scanning microscopy for cellular uptake and cell viability, as well as a facile bioconjugation with a water-soluble UV-active dye as a model cell-targeting biomolecule.
Introduction
Polymer-based drug delivery systems (PDDS) are a promising platform in biomedical applications such as tissue engineering, biomedical implants, bionanotechnology, and drug delivery. Drug molecules are either physically encapsulated inside particles or covalently attached to polymeric chains, thus enhancing therapeutic efficacy while reducing side effects common to small drugs.1–5 Typical examples of PDDS include drug conjugates (prodrugs),6–8 hydrophobic particulates,9–11 vesicles,12 nanocapsules,13,14 and self-assembled micellar aggregates.15–17 Particularly, nanogels are crosslinked hydrogel particles that are confined to nanometer-sized dimensions, which are smaller than micron-sized hydrogel particles (i.e. microgels).18–21 They have the unique properties of hydrogels including high water content, biocompatibility, and adjustable chemical and mechanical properties. These nanogels also possess tunable size, a large surface area for multivalent bioconjugation, and an interior network for the incorporation of therapeutics.22–26Biopolymer-based nanogels (called bionanogels) have all the properties of synthetic counterparts (i.e. nanogels) as well as all benefits from biopolymers being intrinsically biodegradable, abundant in nature, renewable, nontoxic, and relatively cheap.27–30 They also possess a high content of functional groups including hydroxyl, amino, and carboxylic acid groups. These functional groups can be further utilized for bioconjugation with cell targeting agents. In addition to bionanogels, the development of biopolymer-based hydrogels,31–33 capsules,34,35 nanofibrous membranes,36 and polyplexes37 have been explored for various biomedical applications. Despite these advances, the controlled/enhanced release from bionanogels remains challenging.
Enhanced and controlled release of encapsulated anticancer therapeutics in targeted cancer cells is a highly desired property in constructing effective PDDS, particularly nanogels/microgels.38–40 A promising method is to incorporate stimuli-responsive properties into nanogels/microgels. In response to external stimuli, these stimuli-responsive materials either undergo volume changes41,42 or are degraded by cleavage of dynamic covalent bonds;43,44 such changes enable the enhanced release of encapsulated cargos. Typical external triggers include temperature,45,46 pH,47–49 light,50,51 and salts,52 as well as enzymatic53–55 and reductive reactions.56–60 Of our interest, low pH-sensitive systems, while stable at physiological pH, are destabilized in acidic conditions, facilitating the release of encapsulated drugs in a controlled manner. Tumor tissue as well as endosomes and lysosomes present slightly acidic pH (5.0–6.5). Reduction-responsive degradable systems typically employ disulfide–thiol chemistry where disulfides are cleaved to the corresponding thiols in response to reductive reactions.61,62 In biological systems, glutathione (GSH reduced form) exhibits the largely different redox potential between intracellular and extracellular compartments; further, GSH exists at elevated concentration in cancer cells (>10 mM).63,64 In the design and development of smart bionanogels, surprisingly, the stimuli-responsive drug release has not been extensively studied yet. Most stimuli-responsive bionanogels were formed through self-assembly of amphiphilic biopolymers modified with pendant dynamic linkages.65–68
Several methods utilizing chemical crosslinking have been proposed to synthesize various bionanogels in aqueous solution. Typical methods include free radical polymerization (FRP) of methacrylate-modified polysaccharides69,70 and polycondensation/or polyaddition of polysaccharides bearing reactive functional groups.71–74 A promising method involves aqueous FRP of methacrylate monomers in the presence of polysaccharides that allows for the synthesis of polymethacrylate-grafted polysaccharides. This method is versatile in that the properties of polysaccharides can be tuned with varying polymethacrylates. Examples of grafted polymethacrylates include poly(acrylic acids) for pH response,75 as well as poly(N-isopropyl acrylamide)76 and poly(acrylamide)77 for temperature response. However, only few reports describe the synthesis of microgels or nanogels utilizing the aqueous FRP in the presence of dimethacrylate crosslinkers.78,79
This paper describes dual-stimuli reduction and low pH-responsive bionanogels crosslinked with disulfide linkages (ssBNGs) as intracellular delivery nanocarriers with enhanced release. Scheme 1 illustrates our approach. Aqueous FRP of oligo(ethylene oxide) (OEO)-containing methacrylate (OEOMA) in the presence of carboxymethyl cellulose (CMC) was examined to synthesize POEOMA-grafted CMC (POEOMA-g-CMC). Water-soluble POEOMA was targeted to be grafted from CMC as the scaffold materials. POEOMA is an analog of poly(ethylene oxide) (PEO); PEO is biocompatible material that has been FDA-approved for clinical use, has low toxicity, and prevents nonspecific protein adsorption.80,81 An introduction of a disulfide-labeled dimethacrylate (ssDMA) as a crosslinker into aqueous free radical crosslinking polymerization (FRCP) yielded ssBNGs based on disulfide-crosslinked POEOMA-g-CMC. These ssBNGs exhibit dual-controlled release of encapsulated anticancer drugs: reductive cleavage of disulfide crosslinks and acidic pH-response of carboxylic acid (COOH) groups in CMC. Their applicability as intracellular anticancer drug delivery nanocarriers was evaluated using confocal laser scanning microscopy (CLSM) for cellular uptake and cell viability measurements. Further, their facile bioconjugation with a water-soluble UV-active dye as a model cell-targeting biomolecule was demonstrated.
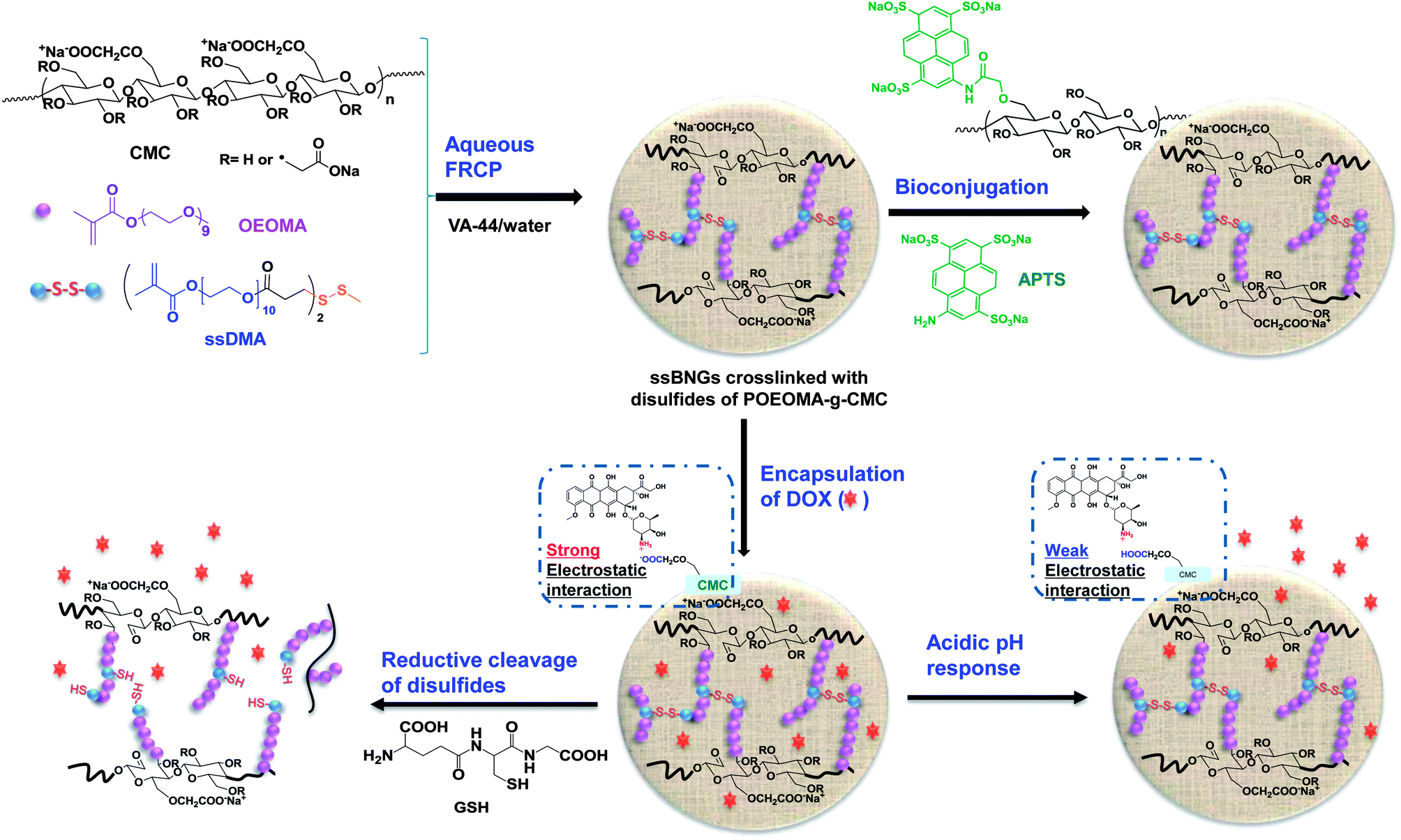 | ||
| Scheme 1 Synthesis by aqueous free radical crosslinking polymerization (FRCP), bioconjugation, and dual stimuli-response of biopolymer-based ssBNGs crosslinked with disulfide linkages of POEOMA-g-CMC. | ||
Experimental
Instrumentation and analysis
1H-NMR spectra were recorded using a 500 MHz Varian spectrometer. The D2O singlet at 4.8 ppm and the CDCl3 singlet at 7.26 ppm were selected as reference standards. Spectral features are tabulated in the following order: chemical shift (ppm); multiplicity (s, singlet; d, doublet; t, triplet; m, complex multiple); number of protons; position of protons. Conversion was determined by 1H-NMR. Molecular weight and molecular weight distribution were determined by gel permeation chromatography (GPC). An Agilent GPC was equipped with a 1260 Infinity Isocratic Pump and a refractive index (RI) detector. Two Agilent PLgel mixed-C and mixed-D columns were used with DMF containing 0.1 mol% LiBr at 50 °C at a flow rate of 1.0 mL min−1. Linear poly(methyl methacrylate) (PMMA) standards from Fluka were used for calibration. Aliquots of polymer samples were dissolved in DMF/LiBr. The clear solutions were filtered using a 0.25 μm PTFE filter to remove any DMF-insoluble species. A drop of anisole was added as a flow rate marker. UV/Vis spectra were recorded on Agilent Cary 60 UV/Vis spectrometer equipped with an external probe.FT-IR measurements
FT-IR spectra of all the samples were taken on a Nicolet 6700 FT-IR spectrometer using KBr pellets. All spectra were recorded with 32 scans in resolution of 4 cm−1 at room temperature in the range of 500–4000 cm−1. Background noises were corrected with pure KBr. For data processing, the baselines of all spectra were corrected. To prepare specimens, as-synthesized aqueous polymer solutions (≈15 mL) were precipitated from acetone (300 mL) under stirring for 12 h. The resulting mixtures were centrifuged under the conditions of 6000 rpm × 10 min × 4 °C. The precipitated polymers were dried in a vacuum oven at 50 °C for 1 day. The dried polymer samples were then mixed with KBr powders to prepare pellets for FT-IR measurements.Dynamic light scattering (DLS) measurements
The sizes in hydrodynamic diameters by volume of POEOMA-g-CMC polymers and ssBNGs in aqueous solution were measured by dynamic light scattering (DLS) at a fixed scattering angle of 173° at 20 °C with a Malvern Instruments Nano S ZEN1600 equipped with a 633 nm He–Ne gas laser.
and

where di is a diameter of particle i and Vi% is a volume faction of particle i. Note that volume faction <0.5% was not included in the calculation.
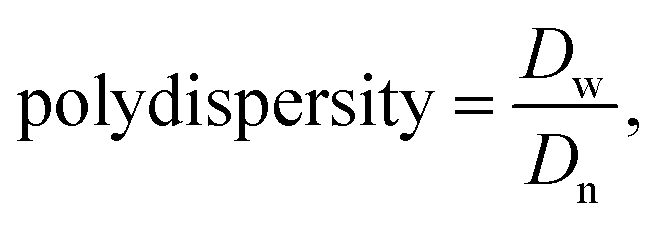
Transmission electron microscopy (TEM)
TEM images were taken using a Philips Tecnai 12 TEM, operated at 120 kV and equipped with a thermionic LaB6 filament. An AMT V601 DVC camera with point to point resolution of 0.34 nm and line resolution of 0.2 nm was used to capture images at 2048 by 2048 pixels. To prepare specimens, the nanogel dispersions were dropped onto copper TEM grids (400 mesh, carbon boated), blotted and then allowed to air dry at room temperature.Materials
Carboxymethylcellulose sodium salts (CMC) with average MW ≈ 250 kg mol−1 and viscosity = 400–800 cP at 2 wt% in water, oligo(ethylene oxide) methacrylate (OEOMA–OH) with MW = 526 g mol−1 and #EO units ≈ 10, 3,3′-dithiopropionic acid (ssDCOOH, 99%), N,N′-dicyclohexyl carbodiimide (DCC), N,N-dimethylaminopyridine (DMAP), doxorubicin hydrochloride (DOX, –NH3+Cl− salt forms, >98%), glutathione (GSH, reduced form), potassium hydrogen phthalate (KHP), and KBr (FT-IR grade, ≥99%) from Aldrich Canada, 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-44) from Wako Chemie, and 8-aminopyrene-1,3,6-trisulfonic acid trisodium salt (APTS, MW = 523.4 g mol−1) from Biotium were purchased and used as received. Dialysis tubing with MWCO = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 with a diameter = 25 mm was purchased from Spectrum Laboratories. Oligo(ethylene oxide) monomethyl ether methacrylate (OEOMA) with MW = 475 g mol−1 and #EO units ≈ 9 was purchased from Aldrich Canada and purified by passing through a column filled with basic alumina to remove inhibitors before used.
000 g mol−1 with a diameter = 25 mm was purchased from Spectrum Laboratories. Oligo(ethylene oxide) monomethyl ether methacrylate (OEOMA) with MW = 475 g mol−1 and #EO units ≈ 9 was purchased from Aldrich Canada and purified by passing through a column filled with basic alumina to remove inhibitors before used.
Dithiopropionyl poly(ethylene glycol) dimethacrylate (ssDMA) was synthesized as described in our previous report.82 Briefly, ssDCOOH (4.6 g, 20 mmol) dissolved in THF (60 mL) reacted with OEOMA–OH (20 g, 38 mmol) in the presence of DCC (7.8 g, 38 mmol) and a catalytic amount of DMAP in dichloromethane (DCM, 120 mL). The product was purified by vacuum filtration and solvent evaporation. 1H-NMR (CDCl3, 7.26 ppm): 1.9 (s, 6H, –CH3), 2.7 (t, 4H, –C(O)–CH2–CH2–SS–), 2.9 (t, 4H, –C(O)–CH2–CH2–SS–), 3.5–3.8 (m, EO protons), 4.0–4.3 (m, 8H, –C(O)O–CH2– and –CH2–O(O)C–), 5.6 (s, 2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), and 6.1 (s, 2H, CH).
), and 6.1 (s, 2H, CH).
Synthesis of ssBNGs crosslinked with disulfides
OEOMA (50 mg, 0.11 mmol), CMC (50 mg), and an aqueous stock solution of ssDMA (0.5 mL, 2 mg mL−1, 0.82 μmol) were dissolved in water (15 mL) in 25 mL of Schlenk flask. The resulting clear mixture was purged with nitrogen for 30 min under magnetic stirring and then heated to 40 °C in a water bath for 10 min. A nitrogen-prepurged aqueous stock solution of VA-44 (0.3 mL, 10 mg mL−1) was added using a syringe to initiate the polymerization. The polymerization was stopped at 5 h by cooling down to room temperature. The resulting ssBNGs were purified by intensive dialysis for 24 h against water for DLS and conjugation experiments as well as against PBS for cell culture experiments.Loading of DOX
DOX (2.3 mg, 5 wt% of ssBNG solids) was dissolved in aqueous ssBNGs solution (5.9 mg mL−1, 8 mL). The resulting mixture was stirred at room temperature for 24 h and dialyzed against water (1 L). Outer water was changed every 12 h to remove free DOX for 24 h, yielding aqueous DOX-loaded ssBNG solution at 4.3 mg mL−1. Their UV/Vis spectra were recorded and the loading level of DOX was calculated by the weight ratio of loaded DOX to dried polymers.Dual-responsive release of DOX using UV/Vis spectroscopy
For acidic pH-responsive release, aliquots of aqueous DOX-loaded ssBNG solution (2 mL) were placed in a dialysis tubing and immersed in aqueous KHP buffer solutions at different pH values (50 mL). The absorbance of DOX in outer water was recorded at an interval of 5 min using a UV/Vis spectrometer equipped with an external probe at λex = 497 nm. For quantitative analysis, the solution in the tubing was combined with outer water and the UV/Vis spectrum of the resulting mixture was recorded. For reduction-responsive release of DOX, the similar procedure was applied except for the use of outer aqueous buffer solution with and without 5 mM GSH at pH = 7.Cell culture
HeLa cancer cells were cultured in DMEM (Dulbecco's modified Eagle's medium) containing 10% FBS (fetal bovine serum) and 1% antibiotics (50 units per mL penicillin and 50 units per mL streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2.Confocal laser scanning microscopy (CLSM)
Cells plated at 2 × 105 cells per well into a 6-well plate were incubated with DOX-loaded ssBNGs (200 μL for DOX = 21 μg) and free DOX (21 μg) as a control at 37 °C for 2 and 8 h. After culture medium was removed, cells were washed with PBS three times. After the removal of supernatants, the cells were fixed with cold methanol (−20 °C) for 20 min at 4 °C. The slides were rinsed with tris-buffered saline Tween-20 (TBST) three times. Cells were stained with 2-(4-amidinophenl)-6-indolecarbamidine (DAPI) for 5 min. The fluorescence images were obtained using a LSM 510 Meta/Axiovert 200 (Carl Zeiss, Jena, Germany).Cell viability using MTT assay
Cells were plated at 5 × 105 cells per well into a 96-well plate and incubated for 24 h in DMEM (100 μL) containing 10% FBS for the following experiments. They were then incubated with various concentrations of empty and DOX-loaded ssBNGs for 48 h. Blank controls (cells only) were run simultaneously. Cell viability was measured using CellTiter 96 Non-Radioactive Cell Proliferation Assay kit (MTT, Promega) according to manufacturer's instruction. Briefly, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solutions (15 μL) was added into each well. After 4 h incubation, the medium containing unreacted MTT was carefully removed. DMSO (100 μL) was added into each well in order to dissolve the formed formazan blue crystals, and then the absorbance at λ = 570 nm was recorded using Powerwave HT Microplate Reader (Bio-Tek). Each concentration was 12-replicated. Cell viability was calculated as the percent ratio of absorbance of mixtures with nanogels to control (cells only).Conjugation of ssBNGs with APTS
An aqueous APTS stock solution (0.83 mL, 5 mg mL−1) was mixed with aqueous ssBNG solution (1 mL, 6 mg mL−1) in the presence of an aqueous stock solution of EDC (1.1 mL, 10 mg mL−1) under stirring at room temperature for 1 day. The mixture was then dialyzed against water for 3 days to completely remove unreacted APTS dyes, yielding aqueous APTS-conjugated ssBNG solution at 1.1 mg mL−1.Results and discussion
Preparation of ssBNGs based on crosslinked POEOMA-g-CMC
Our approach to synthesize dual stimuli-responsive ssBNGs involves the aqueous FRP of OEOMA in the presence of CMC and a disulfide-labeled dimethacrylate (ssDMA). The synthesis of ssDMA using a facile carbodiimide coupling reaction is reported.82 A water-soluble VA-44 azo-type free-radical initiator was selected because of its short half-life at lower temperature (τ1/2 = 10 h at 44 °C). For FRP of OEOMA initiated with VA-44, conversion reached >80% within 2 h at 40 °C (see ESI and Table S1†). Due to the high molecular weight of CMC, the viscosity of its aqueous solution strongly relies on the concentration of CMC. It was found that 1 wt% aqueous CMC solution is suitable for FRP. For the concentration of OEOMA, a series of aqueous FRP of different amounts of OEOMA in the presence of 1 wt% CMC without ssDMA was carried out. The results show that the average diameter of the resulting particles based on POEOMA-grafted CMC increased with an increasing amount of OEOMA (see ESI, Fig. S1 and Table S2†). Such increase is attributed to an increase in probability to intra- or inter-particle coupling reaction being decreased with a decreasing concentration of OEOMA. At 3.3 mg mL−1 concentration, the resulting particles based on POEOMA-grafted CMC had an average diameter ≈14 nm with a monomodal distribution.A 1.5 mol% of ssDMA was introduced into aqueous FRP of OEOMA in the presence of CMC = 1 wt% and VA-44 = 2 wt% of OEOMA at 3.3 mg mL−1. After 5 h, monomer conversion was determined by 1H-NMR to be >80%. The resulting ssBNGs were then purified by intensive dialysis to remove unreacted monomers. DLS results suggest the presence of two populations. The main population (>90% volume) is smaller-sized aggregates with Dn ≈ 24 nm, which is larger than that (≈14 nm) prepared in the absence of ssDMA. In addition, a smaller population (<10% volume) is larger-sized aggregates with the diameter >0.8 μm (Fig. 1a). TEM images indicate near-spherical shapes of ssBNG with average diameter = 19.5 ± 4.1 nm, which is similar to that determined by DLS (Fig. 1b).
 | ||
| Fig. 1 DLS diagram (a) and TEM image (b) of dialyzed ssBNGs. | ||
Aqueous FRP process allows for the synthesis of polymethacrylates grafted from polysaccharide-based biopolymers.83,84 However, this process also forms undesired polymethacrylate homopolymers; such as polyacrylamide,77 poly(methyl methacrylate),85 and poly(3-dimethyl-(methacryloyloxyethyl) ammonium propane sulfonate).86 They were removed by intensive extraction with appropriate solvents.
In our control experiment without ssDMA, the results from our solubility tests suggest that acetone and a mixture of ethanol and acetic acid could be good solvents to POEOMA homopolymers, but poor solvents to CMC (Table S3†). Accordingly, the POEOMA-g-CMC prepared without ssDMA (FRP-C1) was precipitated from acetone to remove undesired POEOMA homopolymers and unreacted OEOMA. As seen in Fig. S2,† the FT-IR spectrum of the POEOMA-g-CMC exhibits two characteristic bands at 1605 cm−1 corresponding to carboxylate (C(O)O−) anion absorption of CMC and at 1730 cm−1 corresponding to ester CO vibrational absorption of POEOMA, suggesting the formation of POEOMA-g-CMC. Similarly, ssBNGs prepared in the presence of ssDMA were precipitated from acetone. Note that the undesired products of POEOMA homopolymers crosslinked in the presence of ssDMA could be dissolved in acetone as microgel particles and thus they will be removed. As seen in Fig. 2, the FR-IR spectrum of the precipitated ssBNGs exhibits the two typical vibrational absorptions at 1605 and 1730 cm−1; this is similar to POEOMA-g-CMC prepared without ssDMA.
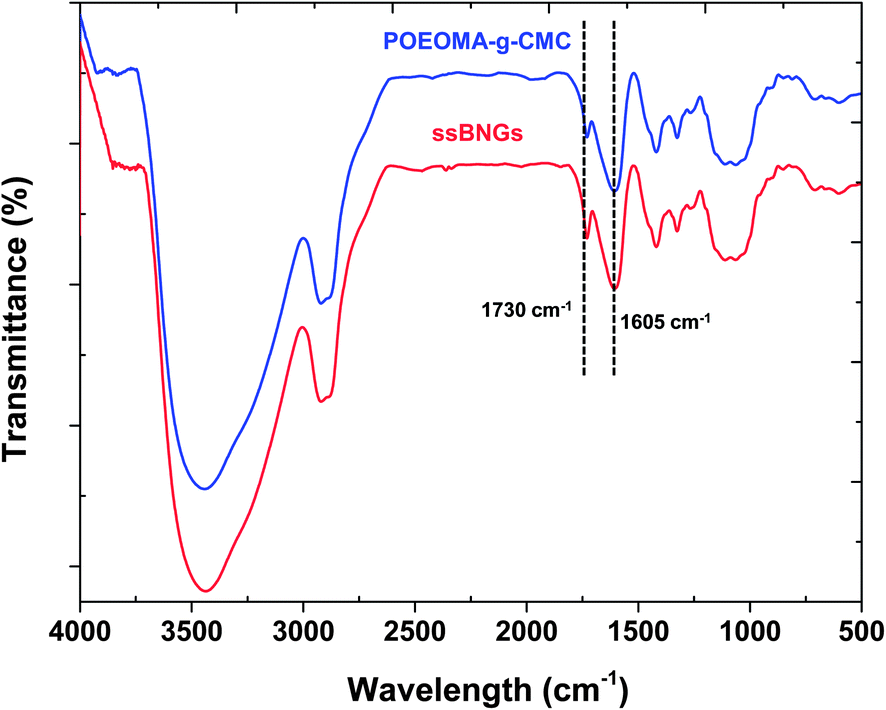 | ||
| Fig. 2 FT-IR spectrum of ssBNGs, compared with that of POEOMA-g-CMC. | ||
Loading of anticancer drugs
To preliminarily evaluate ssBNGs as a platform with enhanced release for tumor-targeting drug delivery, doxorubicin (DOX), a DNA-interacting anticancer drug used in chemotherapy, was encapsulated in ssBNGs. An aqueous DOX stock solution was mixed with ssBNGs in water (pH = 6.5). Free (not encapsulated) DOX was removed by intensive dialysis for 36 h. The removal of free DOX was monitored by measuring the absorbance of DOX at 497 nm in outer water (Fig. S3†). In this way, DOX-loaded ssBNGs in aqueous solution at 4.3 mg mL−1 was prepared. The loading level of DOX for DOX-loaded ssBNGs was determined using UV/Vis spectroscopy. UV/Vis spectrum of DOX-loaded ssBNGs is similar to that of free DOX, suggesting no signification change in the structure of DOX encapsulated in ssBNGs (Fig. S4†). The Beer–Lambert equation with the absorbance and the extinction coefficient of 10000 M−1 cm−1 at λ = 497 nm87 was used to determine the loading level of DOX to be 3.4 ± 0.1% and its loading efficiency to be 68.6 ± 3.1% at the weight ratio of DOX/ssBNGs = 1/20.
Dual responsive release of encapsulated anticancer drugs
The release of DOX from DOX-loaded ssBNGs in response to both acidic pH and thiol reducing agents was investigated. To examine acidic pH-responsive release, aliquots of DOX-loaded ssBNGs in dialysis tubing were placed in aqueous KHP buffer solutions at different pH values of 3, 5.5, and 7. Fig. 3 shows the significant dependence of DOX release upon acidic pH values. Fig. 3b–e show the digital photos of outer water and the corresponding dialysis tubing before and after release. The release of DOX was enhanced in the order of pH 3 > pH 5.5 > pH 7. This enhancement is attributed to the extent of interactions between COOH groups of CMC in ssBNGs and amine (NH2) groups of DOX. Because of pKa of DOX = 8.3, most amine groups of DOX exist as protonated forms (NH3+ forms) in the range of pH values examined here. However, COOH groups exist as their neutral forms (COOH forms) at below pKa = 4.8 or their deprotonated forms (COO− forms) at above pKa. It is obvious that protonated NH3+ forms of DOX have weaker interactions with neutral COOH forms of CMC than deprotonated COO− forms. As a consequence, the release of DOX was faster at pH = 3 than pH = 7. | ||
| Fig. 3 Release of DOX from DOX-loaded ssBNGs in aqueous buffer solutions at different pH of 3, 5.5, and 7 (a), and digital photos of outer water (upper) and the corresponding dialysis tubing (lower) before (b) and after release of DOX at pH = 3 (c), 5.5 (d), and 7 (e). | ||
Next, reductively responsive release of DOX was examined at pH = 7 in a way that aliquots of DOX-loaded ssBNGs in dialysis tubing was placed in aqueous buffer solution with and without (control) 5 mM GSH. As seen in Fig. 4, the release of DOX was relatively faster in the presence of 5 mM GSH, compared to no GSH. Such slight enhancement is the result of GSH-induced degradation of ssBNGs by cleavage of disulfides. These results suggest that acidic pH-response is more significant than thiol-response in enhancing the release of DOX from ssBNGs crosslinked with disulfide linkages of POEOMA-g-CMC.
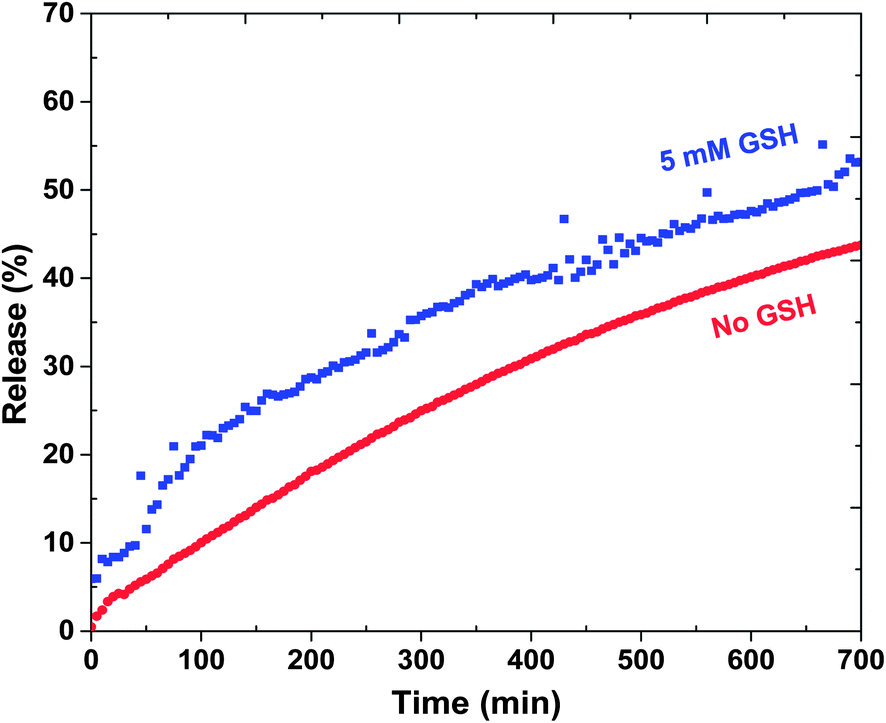 | ||
| Fig. 4 Release of DOX from DOX-loaded ssBNGs in aqueous buffer solutions at pH = 7 with and without 5 mM GSH. | ||
Intracellular release as anticancer drug delivery nanocarriers
Intracellular trafficking of DOX from DOX-loaded ssBNGs after cellular uptake was examined using CLSM. Fig. 5 shows CLSM images of HeLa cells incubated with and without DOX-loaded ssBNGs for 2 and 8 h. For comparison, HeLa cells were also incubated with free DOX. HeLa nuclei were stained with DAPI to provide enhanced contrast and clarity. Compared to free DOX as a control (Fig. 5d and e), the images from DOX fluorescence suggest that DOX-loaded ssBNGs were internalized and DOX was released to reach cell nuclei within 2 h (Fig. 5b). When the incubation time increased to 8 h, the DOX fluorescence became brighter, suggesting more DOX reached cell nuclei (Fig. 5c). | ||
| Fig. 5 CLSM images of HeLa cells incubated with empty ssBNGs for 8 h (a), DOX-loaded ssBNGs for 2 (b) and 8 h (c), as well as free DOX for 2 (d) and 8 h (e). Scale bar = 20 μm. | ||
A MTT colorimetric assay was used to evaluate cytotoxicity of HeLa cells in the presence of empty and DOX-loaded ssBNGs (Fig. 6). The empty crosslinked ssBNGs exhibited >90% HeLa cell viability at concentrations up to 200 μg mL−1; suggesting non-toxicity of ssBNGs to cells. In the presence of DOX-loaded ssBNGs at 200 μg mL−1 (≈3.5 μg mL−1 DOX), however, the viability decreased to below 20%, suggesting inhibition of cell proliferation in the presence of DOX. The results from cell viability and CLSM measurements suggest that DOX release from DOX-loaded ssBNGs, possibly triggered by intracellular GSH and acidic pH inside cancer cells, enhanced the inhibition of the cellular proliferation.
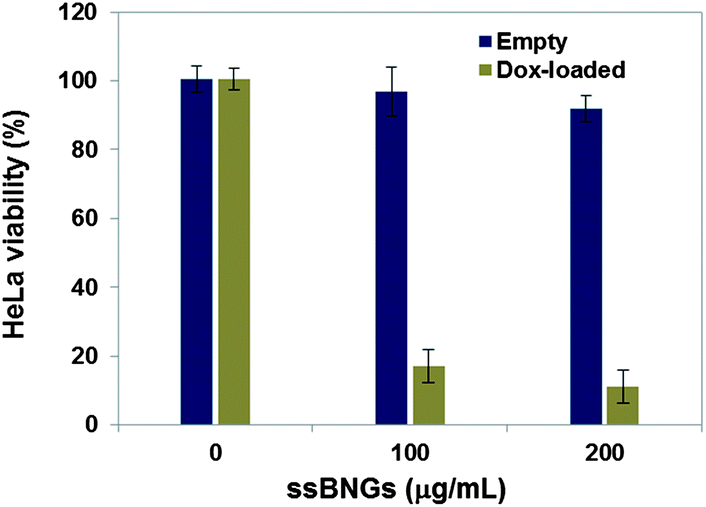 | ||
| Fig. 6 Viability of HeLa cells incubated with empty and DOX-loaded ssBNGs for 48 h determined by MTT assay. Data are presented as the average ± standard deviation (n = 12). | ||
Bioconjugation for active targeting
Active targeting to specific cells through bioconjugation of delivery vehicles with cell targeting agents is a desired property for PDDS. Specific targeting could reduce the serious side effects of drugs as well as enhance the drug efficiency.88,89 Polysaccharides including CMC possess a high content of functional groups including hydroxyl and carboxylic acid groups. These functional groups can be further utilized for bioconjugation with cell targeting agents. To assess an applicability of ssBNGs based on crosslinked POEOMA-g-CMC toward targeted delivery, the conjugation of COOH groups in CMC with a water-soluble APTS dye was examined in aqueous solution through an EDC-coupling reaction.A known amount of ssBNGs was mixed with free APTS in the presence of EDC at room temperature for 1 day. The mole ratio of [COOH]/[APTS]/[EDC] = 1/0.5/4.5. The mole of COOH was calculated from the carboxylate content = 70% (from supplier's catalog). Unreacted APTS dyes were removed by intensive dialysis against water. Fig. 7 shows the UV/Vis spectra of APTS-conjugated ssBNGs and free APTS in aqueous solutions at pH = 7. The maximum absorption at λmax = 424 nm for free APTS dyes was shifted to λmax = 372 nm for APTS-bound ssBNGs. As a control experiment, the pH-dependence of UV absorption of free APTS was examined in aqueous solutions at a broad range of pH from 2 to 10 (Fig. S5†). At above pH = 2, its UV/Vis spectra retained similar with its maximum absorption at λ = 424 nm, while it was blue-shift at below pH = 2. Note that the UV/Vis spectrum of APTS-conjugated ssBNGs was recorded at pH = 7. Consequently, the blue-shift after conjugation is attributed to the formation of new amide linkages, suggesting the successful conjugation of ssBNGs with APTS to form APTS-conjugated ssBNGs (see Scheme 1).
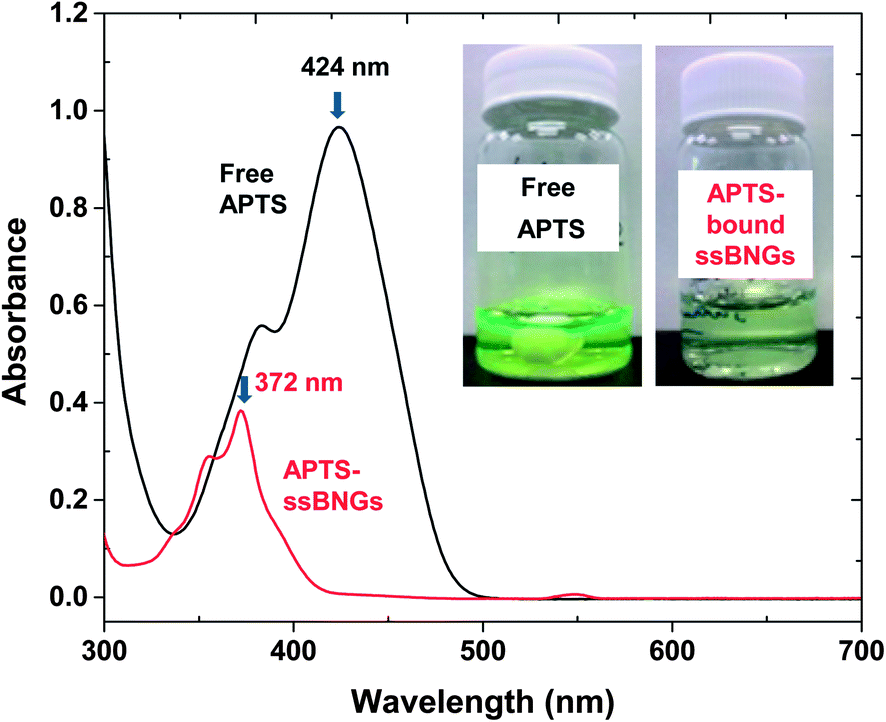 | ||
| Fig. 7 UV/Vis spectra and digital photos (insect) of aqueous solution of APTS-conjugated ssBNGs and free APTS at pH = 7. | ||
For quantitative analysis, the extinction coefficient of APTS at λmax = 424 nm was determined to be ε = 21700 M−1 cm−1 in water (pH = 6.5) (Fig. S6†). UV/Vis spectra of the mixture consisting of ssBNGs, APTS, and EDC before and after the occurrence of EDC coupling reaction are compared in Fig. S7.† Using the extinction coefficient and the difference of the absorbance at λ = 424 nm, the conjugation efficiency of COOH groups in ssBNGs with APTS was estimated to be 15% under these conditions.
Conclusion
Polysaccharide-based ssBNGs crosslinked with disulfide linkages of POEOMA-g-CMC exhibiting dual-stimuli reduction and acidic pH responses were synthesized by a facile aqueous free radical crosslinking polymerization of OEOMA in the presence of CMC and ssDMA at ambient temperature. In the absence of crosslinkers, the initiator concentration, temperature, and OEOMA concentrations were revealed to be important parameters that significantly influence the polymerization kinetics and grafting of POEOMA from CMC chains. Introduction of ssDMA yielded dual stimuli-responsive ssBNGs with diameter <25 nm, confirmed by DLS and TEM. Toward intracellular anticancer drug delivery, DOX was encapsulated at loading level = 3.5%. The results from UV absorbance measurements suggest the enhanced release of DOX in response to acidic pH as well as in the presence of GSH; interestingly, acidic pH-response is more significant than GSH-response in enhancing DOX release. Further, the intracellular release of anticancer drugs after internalization into HeLa cancer cells is confirmed with the results from CLSM and MTT viability assay. These results, combined with the ability to facile bioconjugation with a cell-targeting model biomolecule in aqueous solution, suggest that the ssBNGs are a promising intracellular nanocarrier platform exhibiting multi-controlled drug release.Acknowledgements
Financial supports from Canada Research Chair (CRC) Award are greatly acknowledged. JKO is CRC Tier II in Nanobioscience and a member of the FQRNT-funded Centre Québécois sur les Matériaux Fonctionnels (CQMF). Authors thank Dr N. Chan for TEM measurement and Prof. H. Muchall for our access to FT-IR spectrometer, as well as S. Aleksanian and S. Y. An for helpful discussions.References
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS PubMed.
- R. Langer, Acc. Chem. Res., 2000, 33, 94 CrossRef CAS PubMed.
- A. S. Hoffman, P. S. Stayton, O. Press, N. Murthy, C. A. Lackey, C. Cheung, F. Black, J. Campbell, N. Fausto, T. R. Kyriakides and P. Bornstein, Polym. Adv. Technol., 2002, 13, 992 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3 CrossRef CAS.
- N. A. Peppas, Adv. Drug Delivery Rev., 2004, 56, 1529 CrossRef CAS PubMed.
- J. Khandare and T. Minko, Prog. Polym. Sci., 2006, 31, 359 CrossRef CAS PubMed.
- M. J. Vicent, F. Greco, R. I. Nicholson, A. Paul, P. C. Griffiths and R. Duncan, Angew. Chem., Int. Ed., 2005, 44, 4061 CrossRef CAS PubMed.
- S. Liu, R. Maheshwari and K. L. Kiick, Macromolecules, 2009, 42, 3 CrossRef CAS PubMed.
- O. C. Farokhzad, J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6315 CrossRef CAS PubMed.
- S. H. Kim, J. H. Jeong, K. W. Chun and T. G. Park, Langmuir, 2005, 21, 8852 CrossRef CAS PubMed.
- A. E. van der Ende, E. J. Kravitz and E. Harth, J. Am. Chem. Soc., 2008, 130, 8706 CrossRef CAS PubMed.
- J. Wu and A. Eisenberg, J. Am. Chem. Soc., 2006, 128, 2880 CrossRef CAS PubMed.
- W. Li, J. A. Yoon and K. Matyjaszewski, J. Am. Chem. Soc., 2010, 132, 7823 CrossRef CAS PubMed.
- E. Kim, D. Kim, H. Jung, J. Lee, S. Paul, N. Selvapalam, Y. Yang, N. Lim, C. G. Park and K. Kim, Angew. Chem., Int. Ed., 2010, 49, 4405 CrossRef CAS PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CrossRef CAS PubMed.
- A. S. Mikhail and C. Allen, J. Controlled Release, 2009, 138, 214 CrossRef CAS PubMed.
- X.-B. Xiong, A. Falamarzian, S. M. Garg and A. Lavasanifar, J. Controlled Release, 2011, 155, 248 CrossRef CAS PubMed.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920 CAS.
- A. S. Hoffman, J. Controlled Release, 1987, 6, 297 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307 CrossRef CAS PubMed.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448 CrossRef CAS PubMed.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638 CrossRef CAS PubMed.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707 RSC.
- J. K. Oh, Can. J. Chem., 2010, 88, 173 CrossRef CAS.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418 CrossRef CAS PubMed.
- J. K. Oh, D. I. Lee and J. M. Park, Prog. Polym. Sci., 2009, 34, 1261 CrossRef CAS PubMed.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650 CrossRef CAS PubMed.
- S. A. Agnihotri, N. N. Mallikarjuna and T. M. Aminabhavi, J. Controlled Release, 2004, 100, 5 CrossRef CAS PubMed.
- B. K. Nanjawade, F. V. Manvi and A. S. Manjappa, J. Controlled Release, 2007, 122, 119 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83 CrossRef CAS PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387 CrossRef CAS PubMed.
- E. Hachet, H. Van Den Berghe, E. Bayma, M. R. Block and R. Auzely-Velty, Biomacromolecules, 2012, 13, 1818 CrossRef CAS PubMed.
- P. S. Pramod, K. Takamura, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Biomacromolecules, 2012, 13, 3627 CrossRef CAS PubMed.
- M. A. J. Mazumder, N. A. D. Burke, F. Shen, M. A. Potter and H. D. H. Stover, Biomacromolecules, 2009, 10, 1365 CrossRef CAS PubMed.
- A.-F. Che, Z.-M. Liu, X.-J. Huang, Z.-G. Wang and Z.-K. Xu, Biomacromolecules, 2008, 9, 3397 CrossRef CAS PubMed.
- Y. Wu, P. Ni, M. Zhang and X. Zhu, Soft Matter, 2010, 6, 3751 RSC.
- D. Klinger and K. Landfester, Polymer, 2012, 53, 5209 CrossRef CAS PubMed.
- C. Tsitsilianis, Soft Matter, 2010, 6, 2372 RSC.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174 CrossRef CAS PubMed.
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962 CrossRef CAS PubMed.
- A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088 CrossRef CAS PubMed.
- Q. Zhang, N. R. Ko and J. K. Oh, Chem. Commun., 2012, 48, 7542 RSC.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van Nostrum, J. Controlled Release, 2007, 120, 131 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- S. Aoshima and S. Kanaoka, Adv. Polym. Sci., 2008, 210, 169 CrossRef CAS.
- N. Murthy, M. Xu, S. Schuck, J. Kunisawa, N. Shastri and J. M. J. Frechet, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4995 CrossRef CAS PubMed.
- V. Bulmus, Y. Chan, Q. Nguyen and H. L. Tran, Macromol. Biosci., 2007, 7, 446 CrossRef CAS PubMed.
- E. Themistou and C. S. Patrickios, Macromolecules, 2007, 40, 5231 CrossRef CAS.
- Y. Zhao, Macromolecules, 2012, 45, 3647 CrossRef CAS.
- M. W. Tibbitt, A. M. Kloxin, L. A. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785 CrossRef CAS.
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2004, 43, 6706 CrossRef CAS PubMed.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217 RSC.
- B. Soontornworajit, J. Zhou, M. T. Shaw, T.-H. Fan and Y. Wang, Chem. Commun., 2010, 46, 1857 RSC.
- S. G. Levesque and M. S. Shoichet, Bioconjugate Chem., 2007, 18, 874 CrossRef CAS PubMed.
- R. A. Petros, P. A. Ropp and J. M. DeSimone, J. Am. Chem. Soc., 2008, 130, 5008 CrossRef CAS PubMed.
- H. A. Aliyar, P. D. Hamilton and N. Ravi, Biomacromolecules, 2005, 6, 204 CrossRef CAS PubMed.
- J. K. Oh, D. J. Siegwart, H.-i. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939 CrossRef CAS PubMed.
- B. Gyarmati, B. Vajna, A. Nemethy, K. Laszlo and A. Szilagyi, Macromol. Biosci., 2013, 13, 633 CrossRef CAS PubMed.
- Y. Wang, J. Nie, B. Chang, Y. Sun and W. Yang, Biomacromolecules, 2013, 14, 3034 CrossRef CAS PubMed.
- C. Li, J. Madsen, S. P. Armes and A. L. Lewis, Angew. Chem., Int. Ed., 2006, 45, 3510 CrossRef CAS PubMed.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087 CrossRef CAS.
- A. Russo, W. DeGraff, N. Friedman and J. B. Mitchell, Cancer Res., 1986, 46, 2845 CAS.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199 CrossRef CAS.
- W. Lv, S. Liu, W. Feng, J. Qi, G. Zhang, F. Zhang and X. Fan, Macromol. Rapid Commun., 2011, 32, 1101 CrossRef CAS PubMed.
- J. Tan, H. Kang, R. Liu, D. Wang, X. Jin, Q. Li and Y. Huang, Polym. Chem., 2011, 2, 672 RSC.
- Y.-L. Li, L. Zhu, Z. Liu, R. Cheng, F. Meng, J.-H. Cui, S.-J. Ji and Z. Zhong, Angew. Chem., Int. Ed., 2009, 48, 9914 CrossRef CAS PubMed.
- W. Chen, M. Zheng, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Biomacromolecules, 2013, 14, 1214 CrossRef CAS PubMed.
- D. Klinger, E. M. Aschenbrenner, C. K. Weiss and K. Landfester, Polym. Chem., 2012, 3, 204 RSC.
- L. Messager, N. Portecop, E. Hachet, V. Lapeyre, I. Pignot-Paintrand, B. Catargi, R. Auzely-Velty and V. Ravaine, J. Mater. Chem. B, 2013, 1, 3369 RSC.
- X. Yang, S. Kootala, J. Hilborn and D. A. Ossipov, Soft Matter, 2011, 7, 7517 RSC.
- A. Zhang, Z. Zhang, F. Shi, J. Ding, C. Xiao, X. Zhuang, C. He, L. Chen and X. Chen, Soft Matter, 2013, 9, 2224 RSC.
- N. Morimoto, X.-P. Qiu, F. M. Winnik and K. Akiyoshi, Macromolecules, 2008, 41, 5985 CrossRef CAS.
- S. A. Bencherif, N. R. Washburn and K. Matyjaszewski, Biomacromolecules, 2009, 10, 2499 CrossRef CAS PubMed.
- M.-H. Hsiao, K.-H. Lin and D.-M. Liu, Soft Matter, 2013, 9, 2458 RSC.
- C.-Y. Chuang, W.-Y. Chiu and T.-M. Don, J. Appl. Polym. Sci., 2011, 120, 1659 CrossRef CAS.
- D. R. Biswal and R. P. Singh, Carbohydr. Polym., 2004, 57, 379 CrossRef CAS PubMed.
- M. F. Leung, J. Zhu, F. W. Harris and P. Li, Macromol. Rapid Commun., 2004, 25, 1819 CrossRef CAS.
- C. Duan, J. Gao, D. Zhang, L. Jia, Y. Liu, D. Zheng, G. Liu, X. Tian, F. Wang and Q. Zhang, Biomacromolecules, 2011, 12, 4335 CrossRef CAS PubMed.
- L. Brannon-Peppas, J. Controlled Release, 2000, 66, 321 CrossRef CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 5578 CrossRef CAS PubMed.
- D. W. Jenkins and S. M. Hudson, Chem. Rev., 2001, 101, 3245 CrossRef CAS PubMed.
- A. A. Berlin and V. N. Kislenko, Prog. Polym. Sci., 1992, 17, 765 CrossRef CAS.
- S. M. Derkaoui, T. Avramoglou, C. Barbaud and D. Letourneur, Biomacromolecules, 2008, 9, 3033 CrossRef CAS PubMed.
- L.-M. Zhang and L.-Q. Chen, J. Appl. Polym. Sci., 2002, 83, 2755 CrossRef CAS.
- J. Andrew MacKay, M. Chen, J. R. McDaniel, W. Liu, A. J. Simnick and A. Chilkoti, Nat. Mater., 2009, 8, 993 CrossRef CAS PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra46072j |
| This journal is © The Royal Society of Chemistry 2014 |