
The application of a cytochrome P450 enzyme eluted from encapsulated biomaterials for the catalysis of enantioselective oxidation†
Abstract
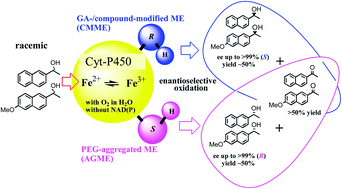
A membrane-bound enzyme (ME) eluted from encapsulated pea protein (PP) under aeration is applicable for the kinetic resolutions of turnover: each enantiomer of rac-1-(6-methoxynaphthalen-2-yl)ethanol (rac-1) can be selectively synthesized (>99% ee; ∼50% chemical yield), utilizing two types of biomaterials: (1) a PEG (1000/4000 = 2/1)-aggregated ME (AGME: synthesizing highly enantiopure R-(−)-1) and (2) a glutaraldehyde (GA)/a PEG (4000)-coated ME (a CMME: synthesizing S-(+)-1 (S-naproxen precursor)). Both reactions occur in the absence of an added cofactor (e.g., NAD(P)) in aqueous media. The specific activities of AGME and CMME were determined to be 0.8 ± 0.03 mU (mean ± SD) mg−1 min−1 and 0.6 ± 0.02 mU (mean ± SD) mg−1 min−1, respectively, and the species exact nature engaged in the key reaction was consistent with that of a heme-binding protein (HBP) based on an N-terminal sequence comparison, which showed 93% similarity with a 20.853 kDa hemophore HasA gene product [Pseudomonas fluorescens Pf-5, a plant commensal bacterium]. The PP-HBP can be regenerated via a successive asymmetric catalytic event using an incorporated iron electron-transfer system in the presence of oxygen—a process seemingly similar to that utilized by the oxygen-driven cytochrome P450 enzyme (Cyt-P450: cysteine – Fe2+ + O2 → Fe3+–O–O− → Fe4+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (oxidizing rac-1 or rac-2) → Fe2+ + H2O). The use of a raw biomaterial as an ME-catalytic system with an incorporated redox cofactor for asymmetric oxidation overcomes the apparent difficulties in working with pure dehydrogenase enzyme/redox cofactor systems for biotransformation.
O (oxidizing rac-1 or rac-2) → Fe2+ + H2O). The use of a raw biomaterial as an ME-catalytic system with an incorporated redox cofactor for asymmetric oxidation overcomes the apparent difficulties in working with pure dehydrogenase enzyme/redox cofactor systems for biotransformation.
 Please wait while we load your content...
Please wait while we load your content...

